Hirschsprungin tauti
Hirschsprungin tauti on polygeeninen perinnöllinen sairaus, joka aiheuttaa muun muassa kroonisen ummetuksen. Hirschsprungin tauti nimettiin tanskalaisen lastenlääkärin Harald Hirschsprungin mukaan.
| Hirschsprungin tauti | |
|---|---|
 |
|
| Luokitus | |
| ICD-10 | Q43.1 |
| Huom! | Tämä artikkeli tarjoaa vain yleistä tietoa aiheesta. Wikipedia ei anna lääketieteellistä neuvontaa. |
Esiintyvyys
Taudin ilmaantuvuus on 1/5 000. [1] Jokaista naista kohden sitä sairastaa 3,9 miestä. Tautia sairastavan sisaruksilla on 200-kertainen riski sairastua tautiin.[2]
Syyt
Hermoston vajaakehitys
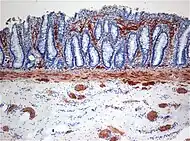
Normaalissa suolistossa suolen poikittaisten ja pitkittäisten lihaskerrosten välissä on Auerbachin hermopunos, joka vastaa suolen peristaltiikasta.[3] Hirschprungin taudissa punos on vajaa paksusuolen ja peräsuolen alueelta, minkä takia kehittymätön osa suolesta on pysyvästi supistunut. [4] Ulostetta alkaa kertyä supistuneen alueen eteen, mikä johtaa lopulta megakooloniin eli paksusuolen laajenemiseen. Sairaus ei välttämättä ole synnynnäinen vaan voi myös aiheutua esimerkiksi Parkinsonin taudin komplikaationa. Kysymyksessä ei aina ole edes itsenäinen sairaus vaan useiden kehitysvammasyndroomien mahdollinen oire.
Synnynnäisessä Hirschprungin taudissa hermostopunoksen vaurio syntyy hermopienan solumigraation aikana raskausviikoilla 5–7. [5]
Genetiikka
Hirschsprungin tautiin vaikuttavia geenejä tunnetaan useita. Geneettiset tekijät vaikuttavat usein sikiökehityksen aikana hermopienan solujen migraatioon ja lisääntymiseen.
RET-geeni koodaa hermopienan solujen reseptoria, ja sen mutaatiot aiheuttavat Hirschprungin tautia. Geenimutaatio on merkittävin tautia aiheuttavista geeneistä, ja se on löydetty 35 %:ssa satunnaisista tautitapauksista. Perinnöllisissä tautitapauksissa mutaatio löytyy noin puolella taudin kantajista.[6] RET-reseptorin ligandi GDNF toimii hyytymistekijänä migroiville soluille. Se ohjaa solut kohti seuraavaksi hermotettavaa suolen aluetta. GDNF muodostaa GFL-GFRα-kompleksin ennen sitoutumistaan RET-reseptoriin. Näin mutaatiot myös GDNF-ligandia tai sen kompleksia koodaavissa geeneissä voivat aiheuttaa Hirschsprungin tautia.[7]
EDN3-ligandin sitoutuessa ENDRB-geenin koodaamaan reseptoriin käynnistyy signaalikaskadi, joka vaikuttaa kohdesolun lisääntymiseen ja erilaistumiseen. Signaalireitillä on tärkeä rooli migraation ylläpidossa, sillä erilaistuneet solut eivät voi edetä suolessa. Liian aikainen erilaistuminen siten hidastaa normaalia migraatiota, joka edelleen aiheuttaa Hirschprungin tautia. Mutaatiot reseptorin, ligandin tai ligandin aktivoivan ECE1-entsyymin geenissä aiheuttavat siten Hirschsprungin tautia.[8][9]
Oireet
Hirschsprungin taudissa paksusuolen ja peräsuolen alueella on hermoston puutteellisuudesta johtuva tukos. Suoleen kerääntyvä uloste aiheuttaa megakooloniksi kutsutun tilan, missä paksunsuolen seinä turpoaa. Uloste ei pääse tukoksen yli, mikä aiheuttaa ummetusta, vatsan pinkeyttä ja oksentelua. [10]
Toteaminen ja hoito
Taudin diagnoosi tehdään yleensä pian syntymän jälkeen. Mikäli vastasyntyneen ensimmäinen uloste, niin kutsuttu lapsenpihka, ei tule 48 tunnin kuluessa syntymästä, on syytä epäillä Hirschsprungin tautia. Taudin todentamiseen voidaan käyttää suolen varjoainekuvausta, ja alustavan diagnoosin jälkeen hermoganglioiden puutos voidaan todeta solunäytteellä.[11] Myös peräaukon sulkijalihaksen refleksiä voidaan käyttää diagnoosin varmentamiseen, kun lapsi on 12 päivää vanha.[9]
Hirschsprungin taudin hoito on yleensä kirurginen. Lyhyessä ja keskilyhessä fenotyypissä tehdään niin kutsuttu TERPT (transanal endorectal pull-through) -leikkaus, jossa vaurioitunut osa suolta vedetään anuksen läpi ja leikataan pois. Mikäli leikkaus ei tuota haluttua tulosta, tautia voidaan hoitaa myös botuliiniruiskeella supistuneelle alueelle.[9]
Lähteet
- http://www.duodecimlehti.fi/web/guest/arkisto?p_p_id=dlehtihaku_view_article_WAR_dlehtihaku&p_p_action=1&p_p_state=maximized&p_p_mode=view&_dlehtihaku_view_article_WAR_dlehtihaku__spage=%2Fportlet_action%2Fdlehtihakuartikkeli%2Fviewarticle%2Faction&_dlehtihaku_view_article_WAR_dlehtihaku_tunnus=duo97245&_dlehtihaku_view_article_WAR_dlehtihaku_p_frompage=uusinnumero
- Qian Jiang, Yen-Yi Ho, Li Hao, Courtney Nichols Berrios, Aravinda Chakravarti: Copy Number Variants in Candidate Genes Are Genetic Modifiers of Hirschsprung Disease. PLoS ONE, 21.6.2011, nro 6, s. e21219. PubMed:21712996. doi:10.1371/journal.pone.0021219. ISSN 1932-6203. Artikkelin verkkoversio. en
- A. T. Rake: Achalasia and Degeneration of Auerbach's Plexus. Proceedings of the Royal Society of Medicine, 1928-9, nro 11, s. 1775–1777. PubMed:19986622. ISSN 0035-9157. Artikkelin verkkoversio.
- H. H. Nixon: HIRSCHSPRUNG'S DISEASE. Archives of Disease in Childhood, 1964-4, nro 39, s. 109–115. PubMed:14131948. ISSN 0003-9888. Artikkelin verkkoversio.
- Alan J. Burns: Migration of neural crest-derived enteric nervous system precursor cells to and within the gastrointestinal tract. The International Journal of Developmental Biology, 2005, nro 2–3, s. 143–150. PubMed:15906227. doi:10.1387/ijdb.041935ab. ISSN 0214-6282. Artikkelin verkkoversio.
- Titis Widowati, Shamiram Melhem, Suryono Y. Patria, Bianca M. de Graaf, Richard J. Sinke, Martijn Viel: RET and EDNRB mutation screening in patients with Hirschsprung disease: Functional studies and its implications for genetic counseling. European journal of human genetics: EJHG, 06 2016, nro 6, s. 823–829. PubMed:26395553. doi:10.1038/ejhg.2015.214. ISSN 1476-5438. Artikkelin verkkoversio.
- H. M. Young, C. J. Hearn, P. G. Farlie, A. J. Canty, P. Q. Thomas, D. F. Newgreen: GDNF is a chemoattractant for enteric neural cells. Developmental Biology, 15.1.2001, nro 2, s. 503–516. PubMed:11150245. doi:10.1006/dbio.2000.0100. ISSN 0012-1606. Artikkelin verkkoversio.
- Amanda Barlow, Esther de Graaff, Vassilis Pachnis: Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron, 4.12.2003, nro 5, s. 905–916. PubMed:14659090. ISSN 0896-6273. Artikkelin verkkoversio.
- Naomi E. Butler Tjaden, Paul A. Trainor: The developmental etiology and pathogenesis of Hirschsprung disease. Translational Research: The Journal of Laboratory and Clinical Medicine, 2013-7, nro 1, s. 1–15. PubMed:23528997. doi:10.1016/j.trsl.2013.03.001. ISSN 1878-1810. Artikkelin verkkoversio.
- Kustannus Oy Duodecim: Hirschsprungin tauti (Orphanet) Duodecim - Terveyskirjasto. Viitattu 10.11.2018.
- R J Andrassy, H Isaacs, J J Weitzman: Rectal suction biopsy for the diagnosis of Hirschsprung's disease.. Annals of Surgery, 1981-4, nro 4, s. 419–424. PubMed:7212804. ISSN 0003-4932. Artikkelin verkkoversio.