Orbital molekular
Orbital molekularrak, kimika kuantikoan, elektroi-dentsitatea duten espazioko eremuak dira, elektroiek molekuletan izan dezaketen portaera ondulatorioa deskribatzen duten funtzio matematikoen bidez definitzen direnak. Funtzio horiek propietate kimiko eta fisikoak kalkulatzeko erabil daitezke, hala nola espazioko eskualde batean elektroi bat aurkitzeko probabilitatea kalkulatzeko. Orbital terminoa lehen aldiz ingelesez aurkeztu zuen Robert S. Mullikenek 1932an, «elektroi baten uhin-funtzio orbitalaren» laburdura gisa (one-electron orbital wave function[1]). Terminoa 1925ean Erwin Schrödingerrek erabilitako hitz alemanaren itzulpen batetik abiatuta proposatu zuen, 'Eigenfunktion'. Ordutik, funtzio horrekin sortutako espazioaren eskualdearen sinonimotzat hartzen da. Orbital molekularrak eratzeko, eskuarki, molekularen atomo bakoitzean zentratutako orbital atomikoen konbinazio lineala erabiltzen da. Egitura elektronikoa kalkulatzeko metodoak erabiliz, hala nola Hartree-Focken metodoa edo eremu autokonsistenteena (self-enlent field, SCF), modu kuantitatiboan lor daitezke.

Konfigurazio elektronikoa
Orbital molekularrak molekulen konfigurazio elektronikoa zehazteko erabiltzen dira. Honek sistema molekularraren egoera elektronikoa espin-orbitalen produktu antisimetrizatu gisa deskribatzea ahalbidetzen du. Horretarako, orbital molekularrak orbital atomikoen konbinazio lineal gisa adierazi ohi dira[2]. Aplikazio garrantzitsu bat, gutxi gorabeherako orbital molekularrak molekuletako lotura deskribatzeko eredu sinple gisa erabiltzea da.
Kimika kuantikoko metodo gehienak sistemaren orbital molekularrak kalkulatzen hasten dira. Orbital molekularrak nukleoek sortutako eremu elektrikoan elektroi batek duen portaera deskribatzen du, baita gainerako elektroien batez besteko banaketa ere. Orbital berean dauden bi elektroiren kasuan, Pauliren bazterketa printzipioak kontrako spinak izatera behartzen ditu.
Orbital molekularrak kualitatiboki lortzea
Egitura molekularra kualitatiboki deskribatzeko, orbital molekularrak lor daitezke orbital atomikoen konbinazio lineal gisa hurbilduz.
Obital molekularrak kualitatiboki lortzeko, ondorengo oinarrizko arauak jarraitu behar dira.
Lehenengoa, orbital molekularren kopurua hedapen linealean sartutako orbital atomikoen kopuruaren berdina da.
Bigarrena arauaren arabera, orbital atomikoak gehiago nahasten dira (hau da, gehiago laguntzen diete orbital molekular berberei) antzeko energiak badituzte. Hori gertatzen da molekula diatomiko homonuklearren kasuan, hala nola O2aren kasuan. Hala ere, nukleo ezberdinak elkartzen direnean, karga desberdinak (eta, beraz, karga eraginkorrak eta elektronegatibotasunak) orbital molekularra deformatzen du. Hala, hidrogenoaren bi 1s orbitalak %50ean gainjartzen dira, eta bi orbital molekularrak era berdinean eratzen laguntzen dute. HO loturan, berriz, oxigenoak parte-hartze handiagoa du, eta orbital molekularrak oxigenoaren orbital atomikoaren antz handiagoa du.
Hirugarrena, orbital atomikoak simetria-arauek baimentzen badute soilik nahasten direla da. Simetria-taldearen irudikapen laburtezin desberdinen arabera aldatzen diren orbitalak ez dira nahasten. Horren ondorioz, ekarpen garrantzitsuenak gehien gainjartzen diren orbital atomikoetatik datoz.
Hidrogeno molekula
Adibide sinple gisa, adierazgarria da H2 dihidrogeno molekula, H' eta H'' etiketadun bi atomorekin. Energia gutxien duten orbital atomikoak, 1s' eta 1s'', ez dira molekularen simetriaren arabera aldatzen. Baina ondorengo konbinazio linealek bai:
| 1s' - 1s" | Konbinazio antisimetrikoa: erreflexioak ukatua, beste ariketen ez dute aldatzen |
| 1s' + 1s" | Konbinazio simetrikoa: Ariketek ez dute aldatzen |
Oro har, konbinazio simetrikoa (orbital lotzailea) energia aldetik txikiagoa da jatorrizko orbitalak baino, eta konbinazio antisimetrikoa (orbital antilotzailea) handiagoa da. H2 dihidrogeno molekulak bi elektroi dituenez, biak orbital lotzaileak deskribatu ditzake, eta, hala, sistemak energia txikiagoa du (beraz, egonkorragoa da) bi hidrogeno aske atomo baino. Horri lotura kobalente deritzo.
Sir John Lennard-Jonesek orbital molekularrak orbital atomikoen konbinazio lineal gisa (OM-CLOA) hurbiltzeko metodoa proposatu zuen 1929an[3]. Difluor eta dioxigeno molekulen egitura elektronikoa printzipio kuantikoetatik nola eratortzen den erakutsi zuen. Orbital molekularren teoriara kuantitatiboki hurbiltze horrek kimika kuantiko modernoaren sorrera ekarri zuen.
Orbital molekular motak
Bi atomo lotzean, orbital atomikoak fusionatu egiten dira orbital molekularrak emateko:
- Lotzaileak: sortu dituen orbital atomikoetako edozeinek baino energia gutxiago dute. Erakarpen-egoeran dago, hau da, nukleoen arteko eremuan. Hala, nukleo positiboek aldarapen-indar elektrostatikoak gainditzen dituzte, haien artean dagoen karga negatiboko hodei elektronikoak eragiten duen erakarpenari esker, lotura-luzera deritzon distantzia jakin bateraino.
- Antilotzaileak: Energia handiagokoak, eta ondorioz, aldarapen egoeran.
Orbital molekular motak hauek dira:
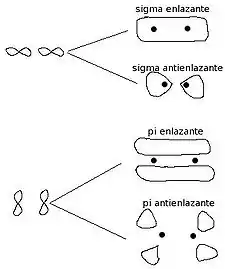
- σ orbital lotzaileak: s orbital atomikoak p orbitalekin konbinatzea (s-s p-p s-p p-s). Deslokalizazio-maila oso txikia duten lotura "errazak". Lotura-ardatzaren inguruan geometria zilindrikoa duten orbitalak.
- π Orbital lotzaileak: lotura-ardatzean perpendikular dauden p orbital atomikoen konbinazioa. Oso deslokalizatutako elektroiak, ingurunearekin erraz elkarreragiten dutenak. Hodei elektroniko gisa banatzen dira lotura-planoaren gainean eta azpian.
- σ* orbital antilotzaileak: lotzaileen bertsio eszitatua (energia handiagokoa).
- π* orbital antilotzaileak: energia handiko π orbitalak.
- n orbitalak: heteroatomoak dituzten molekulentzat (adibidez, N edo O orbitalak). Elektroi desparekatuek ez dute loturan parte hartzen eta orbital hori betetzen dute.
Orbital molekularrak elektroiz "betetzen" dira, orbital atomikoak bezala:
- Energia-mailaren goranzko ordenan: orbital lotzaileak antilotzaileak baino lehen betetzen dira, eta horien artean energia-ordena txikiagoa dutenak lehenik, eta altuagokoak ondoren. Molekulak orbitalak betetzera joko du, energia-egoera ona izan dadin.
- Pauliren esklusio-printzipioari jarraituz: orbital molekularrak eratzen direnean, orbital horiek bi elektroi izan ditzakete gehienez, eta horiek spin desberdinak izango dituzte.
- Hunden biderkatzaile maximoaren araua aplikatuta: endekatutako orbital molekularrek (energia-maila bera dutenek) elektroiak banatzeko joera dute. Hori orbital erdi-beteak lortzeko ematen da, azken hauek egonkorragoak baitira azpigeruza bete eta beste huts bat baino, elektroien arteko aldarapen-indar handiak direla eta. Horri esker, zenbait molekularen propietateak azal daitezke, hala nola oxigeno molekularraren paramagnetismoa (molekularen orbital kanpokoenak elektroi desparekatuak ditu, eremu magnetiko batekin elkarrekintzan ari direnak).
Arau horien arabera, orbitalak osatzen dira. Molekula bat egonkorra izango da elektroiak gehienbat orbital lotzaileetan badaude, eta ezegonkorra izango da orbital antilotzaileetan badaude:
Ikus gainera
Erreferentziak
- Mulliken, Robert S.. (julio de 1932). «Electronic Structures of Polyatomic Molecules and Valence. II. General Considerations» Physical Review 41 (1): 49–71. doi:. Bibcode: 1932PhRv...41...49M..
- Albright, Thomas A.. (2013). Orbital interactions in chemistry. (Second edition. argitaraldia) ISBN 978-0-471-08039-8. PMC 823294395. (Noiz kontsultatua: 2022-12-21).
- (Ingelesez) Lennard-Jones, J. E.. (1929-01-01). «The electronic structure of some diatomic molecules» Transactions of the Faraday Society 25 (0): 668–686. doi:. ISSN 0014-7672. (Noiz kontsultatua: 2022-12-21).
| Autoritate kontrola |
|
|---|
 Datuak: Q725417
Datuak: Q725417 Multimedia: Molecular orbitals / Q725417
Multimedia: Molecular orbitals / Q725417