Urticaria pigmentosa
Die Urticaria pigmentosa ist eine sehr seltene angeborene Erkrankung mit dem Hauptmerkmal einer Urticaria aufgrund einer Mastozytose der Haut.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q82.2 | Mastozytose (angeboren) – Urticaria pigmentosa |
| ICD-10 online (WHO-Version 2019) | |
Synonyme sind: Nettleshipsche Krankheit; Nettleship-Syndrom; Mastozytose-Syndrom; kutane Mastozytose; Braunmensch; Mastzellenretikulose, Morbus Rywlin, generalisierte Mastozytose; englisch Maculopapular cutaneous mastocytosis
Die Namensbezeichnung bezieht sich auf den Autor der Erstbeschreibung aus dem Jahre 1869 durch Edward Nettleship.[3][4]
Die Erkrankung ist nicht zu verwechseln mit dem Nettleship-Falls-Syndrom, einem okulären Albinismus.
Verbreitung
Häufigkeit und der Vererbungsmodus sind nicht bekannt.[1]
Ursache
Der Erkrankung liegen Mutationen im KIT-Gen auf Chromosom 4 Genort q12 zugrunde.[5]
Einteilung
Folgende Unterteilungen ist gebräuchlich:
Systematisch:
Klinisch:[2]
- Hämorrhagische Form (Urticaria pigmentosa haemorrhagica)
- Exanthematische Form (Urticaria pigmentosa, exanthematische)
- Bullöse Form (Urticaria pigmentosa bullosa)
- Xanthelasmoide Form (Urticaria pigmentosa xanthelasmoidea)
Klinische Erscheinungen
Klinische Kriterien sind:[2]
- Erstmanifestation häufig bereits in den ersten 2 Lebensjahren, dann wieder gehäuft zwischen dem 4. und 6. Lebensjahrzehnt
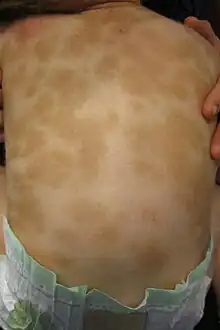
- Lokalisation hauptsächlich am Rumpf
- Flache graubraune Flecken mit Darier-Zeichen, häufig Dermographismus elevatus
Differentialdiagnose
Abzugrenzen ist eine systemische Form der Mastozytose; ferner ein Malignes Lymphom oder ein Dermatofibrom.
Therapie
Neben dem Vermeiden von Auslösern wie u. a. Nichtsteroidale Antiphlogistika, Acetylsalicylsäure, Kodein, Procain, Polymyxin B, mechanische Reizung oder abrupte Temperaturänderungen stehen kühlende oder Antihistaminika-enthaltende Gele sowie die PUVA zur Verfügung.[2]
Heilungsaussicht
Die Prognose ist günstig, die meisten Betroffenen haben eine normale Lebenserwartung. Bei Manifestation im Kindesalter kommt es in gut der Hälfte zu einer Spontanremission.[9]
Literatur
- R. J. Vasani, S. V. Medhekar: Urticaria pigmentosa. In: Indian dermatology online journal, Band 6, Nr. 6, 2015 Nov-Dec, S. 464–465, doi:10.4103/2229-5178.169727, PMID 26752589, PMC 4693376 (freier Volltext).
Weblinks
Einzelnachweise
- Maculopapular cutaneous mastocytosis. In: Orphanet (Datenbank für seltene Krankheiten).
- Enzyklopädie Dermatologie
- E. Nettleship: Rare forms of urticaria. In: British Medical Journal, London 1869, Band 2, S. 323–324.
- E. Nettleship: Chronic urticaria leaving brown stains: nearly two years’ duration. In: British Medical Journal, London 1869, Band 2, A 435
- Mast cell disease. In: Online Mendelian Inheritance in Man. (englisch)
- Urtikaria pigmentosa, noduläre. In: Orphanet (Datenbank für seltene Krankheiten).
- Urtikaria pigmentosa, plaqueförmige. In: Orphanet (Datenbank für seltene Krankheiten).
- Urtikaria pgimentosa, typische. In: Orphanet (Datenbank für seltene Krankheiten).
- A. Heinze, T. J. Kuemmet, Y. E. Chiu, S. S. Galbraith: Longitudinal Study of Pediatric Urticaria Pigmentosa. In: Pediatric dermatology. [elektronische Veröffentlichung vor dem Druck] Januar 2017, doi:10.1111/pde.13066, PMID 28133781.