Proteasom
Der Begriff Proteasom bezeichnet im Allgemeinen einen Proteinkomplex, der in Archaeen und einigen Bakterien vorkommt und bei Eukaryoten sowohl im Zellkern als auch im Zytoplasma vorliegt. Im Speziellen unterscheiden sich Proteasome in ihrer Zusammensetzung zwischen verschiedenen Taxa. In Säugetieren liegen Proteasome auch innerhalb eines Organismus in verschiedenen Formen vor, z. B. als freies 20S Proteasom oder in Kombination mit einem oder mehreren regulatorischen Partikeln (z. B. als 26S Proteasom). Proteasome spielen eine essenzielle Rolle für den kontrollierten Abbau von Proteinen in der Zelle und sind damit integraler Bestandteil der Proteinqualitätskontrolle in Zellen. Zum Abbau werden bestimmte Proteine entfaltet, in das Proteasom eingeschleust und dort von den katalytisch aktiven Untereinheiten des Proteasoms in kürzere Peptide geschnitten. Es handelt sich bei Proteasomen daher um multikatalytische Proteasen.
| Proteasom | ||
|---|---|---|
| Enzymklassifikation | ||
| EC, Kategorie | 3.4.25.1, Peptidase | |
| MEROPS | T1 | |
| Reaktionsart | Proteolyse | |
| Substrat | ubiquitin(yl)ierte Proteine | |
Struktur
Das eukaryotische Proteasom besteht aus dem 20S Kern-Partikel, in dem die katalytischen Proteaseaktivitäten lokalisiert sind, und ggf. weiteren regulatorischen Proteinkomplexen, die an eines oder beide Enden des 20S Proteasoms gebunden sein können. Von besonderer Bedeutung ist der 19S Regulator (auch PA700), der eine zentrale Rolle beim Abbau ubiquitinierter Proteinsubstrate einnimmt. 20S Proteasome in Kombination mit einem oder zwei 19S Regulatoren werden als 26S Proteasom bezeichnet. Die weitere Unterscheidung zwischen 26S (20S + 1 x 19S) und 30S (20S + 2 x 19S) ist in der Literatur uneinheitlich. Neben dem 19S Regulator gibt es weitere Regulatoren wie z. B. den 11S Regulator (PA28αβ oder PA28γ) oder den im Zellkern lokalisierten Regulator PA200[1]. Proteasome können auch aus dem 20S Proteasom mit zwei verschiedenen Regulatoren bestehen, z. B. einem 19S Regulator an einem Ende und einem PA28αβ Regulator am anderen Ende. Diese Strukturen werden als Hybrid-Proteasome bezeichnet. Das 20S Proteasom formt eine zylindrische Struktur aus vier Ringen zu je sieben Untereinheiten. In der Mitte der beiden α-Ringe befindet sich eine Pore (englisch: gate), die den Eintritt von Substraten ins 20S Proteasom reguliert. Die Größe der Öffnung wird vorrangig vom gebundenen Regulator-Komplex reguliert. Die „Gate-Opening“ Funktion des Regulators wurde zuerst für 20S Proteasome der Hefe in Kombination mit PA26 Regulatoren aus dem Protozoon Trypanosoma brucei beschrieben[2]. Zudem wurde auch eine allosterische Regulation zwischen den katalytisch aktiven Untereinheiten des 20S Proteasoms und der α-Ring-Öffnung beschrieben[3].
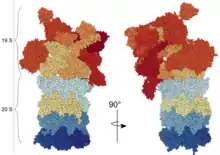


20S Proteasom (auch 20S Kern-Partikel)
Das 20S Proteasom hat die Form eines Zylinders und wirkt als multikatalytische Protease. Es besteht aus vier Ringen, die ihrerseits aus jeweils 7 verschiedenen Untereinheiten zusammengesetzt sind. Die äußeren Ringe bestehen aus α-Untereinheiten (α1-α7). Im Unterschied zu bakteriellen und Archeae-Proteasomen, die mehrere identische katalytische Untereinheiten besitzen, bestehen die inneren zwei Ringe der eukaryotischen Proteasome aus je sieben β-Untereinheiten, von denen jeweils drei Untereinheiten katalytisch aktive Proteasen darstellen (β1, β2 und β5). In der Mitte des 20S Proteasoms wird somit strukturell eine innere Kammer geformt, in der die Spaltung der Substrate abläuft. Zwischen den α- und den β-Ringen ergibt sich strukturell je eine weitere Kammer (Antechamber). Die katalytisch aktiven Untereinheiten kommen bei vielen Wirbeltieren in je zwei (für β1 und β2) oder drei (für β5) verschiedenen Varianten vor und die Zusammensetzung des 20S Proteasoms aus den jeweiligen Varianten bestimmt die Unterscheidung des Proteasoms als Standardproteasom (auch „konstitutives Proteasom“ genannt), Immunproteasom oder Thymoproteasom. In Standardproteasomen werden die Untereinheiten β1, β2 und β5 als Standardproteasom-Untereinheiten (auch konstitutive Untereinheiten) bezeichnet. Jede dieser drei Untereinheiten hat eine etwas andere proteolytische Aktivität: β1 spaltet die Peptidkette des entfalteten Proteins nach sauren Aminosäuren (Caspase-ähnliche Aktivität), β2 spaltet nach basischen Aminosäuren (Trypsin-ähnliche Aktivität) und β5 spaltet nach hydrophoben Aminosäuren (Chymotrypsin-ähnliche Aktivität). Die Struktur des 20S Proteasoms konnte mit hoher Auflösung durch Röntgen-Struktur-Analyse bestimmt werden[5].
19S Regulator
Die 19S Komplexe sitzen bei Eukaryoten ähnlich einer Haube (cap) auf einer Öffnung oder beiden Öffnungen des 20S Komplexes. Sie regulieren den Zugang von Substraten zum 20S Komplex, indem sie Substratproteine entfalten und die Größe der α-Ring-Pore im 20S Komplex regulieren. Der 19S Regulator besteht aus Rpn- und Rpt-Proteinen. Die verschiedenen Rpn Untereinheiten (regulatory particle non-ATPases) sind zuständig für die Bindung von ubiquitinierten Substraten an den Proteasom-Komplex. Sie regulieren die Entfaltung des zum Abbau bestimmten Proteins, rekrutieren andere Faktoren ans Proteasom (z. B. das de-ubiquitinierende Enzym USP14) oder sind selbst an der De-Ubiquitinierung beteiligt (Rpn11)[6]. Am Transfer des Substratproteins in das 20S Proteasom sind die sechs Rpt-Untereinheiten (regulatory particle ATPasen) maßgeblich beteiligt. In Abhängigkeit von Adenosintriphosphat (ATP) und der Hydrolyse von ATP zu ADP durchläuft der Rpt-Ring einen komplexen Komformationszyklus, durch den die Substrate zum Abbau ins 20S Proteasom transloziert werden. Die Struktur des 19S Regulators konnte aufgrund seiner Dynamik erst durch die Entwicklung der Kryo-Elektronenmikroskopie in jüngerer Zeit näher aufgeklärt werden[7][8].
Entdeckung des Proteasoms
Die Entdeckung des Proteasoms geht maßgeblich auf die Dekade der 1980er Jahre zurück. Es wurde ursprünglich unter mehreren verschiedenen Namen unabhängig voneinander beschrieben, darunter auch unter der Bezeichnung Multikatalytischer Protease Komplex,[9] und schließlich Ende der 1980er Jahre unter dem Begriff Proteasom, welcher die Rolle dieses Komplexes für den Abbau von Proteinen anzeigen sollte[10]. In den frühen 1990er Jahren wurden die ersten Gene (Psmb8/LMP7 und Psmb9/LMP2) der Untereinheiten des sogenannten Immunproteasoms entdeckt, deren Position im Gen-Cluster des Haupthistokompatibilitätskomplexes liegt. 1996 wurde die dritte Untereinheit (Psmb10/MECL-1) des Immunproteasoms entdeckt[11][12] und schließlich im Jahre 2007 eine weitere alternative Untereinheit (Psmb11/β5t), die ausschließlich im Thymus vorkommt. Immunproteasome und Thymoproteasome haben sich in der Evolution durch Genduplikationen entwickelt und kommen in den Wirbeltieren der Überklasse Kiefermäuler vor, jedoch anscheinend mit Ausnahme der Vögel[13].
Immunproteasom
Die katalytischen Untereinheiten β1, β2 und β5 werden auch als konstitutive Untereinheiten bezeichnet und das komplette 20S Proteasom mit diesen Untereinheiten als konstitutives Proteasom oder Standard-Proteasom. Diese nähere Spezifikation ist von Bedeutung für die besondere Rolle des Proteasoms im Rahmen einer Immunantwort. Im Verlauf einer solchen werden Zellen an Orten eindringender Pathogene (Viren, Bakterien, Parasiten) durch Zellen des Immunsystems dem Zytokin Interferon-γ ausgesetzt. Dadurch verändern die Zellen die Expression verschiedener Gene, wodurch die Immunantwort unterstützt wird. Anstelle der drei Proteasom-Untereinheiten β1, β2 und β5 werden dann die Untereinheiten Low molecular mass protein 7 (LMP7, auch β5i), Multicatalytic endopeptidase complex like 1 (MECL-1, auch β2i) und LMP2 (auch β1i) in den 20S Kern-Partikel eingebaut. Das so zusammengesetzte Proteasom wird daher als "Immunproteasom" bezeichnet[14]. Das Immunproteasom hat zahlreiche Funktionen für die Immunantwort, beispielsweise eine stärkere Präsentation von Antigenen über MHC-Klasse I im Vergleich zum Standard-Proteasom. Darüber hinaus werden dem Immunproteasom weitere Funktionen zugeschrieben, die unabhängig von der Antigenpräsentation sind, wie z. B. ein Einfluss auf das Überleben von T-Zellen während einer Virus-Infektion oder ein Einfluss auf die funktionale Polarisierung von T-Helferzellen. Eine besondere Rolle des Immunproteasoms für die Protein-Homöostase bei Vorliegen von zellulärem Stress unter dem Einfluss von entzündlichen Zytokinen wie dem Interferon-γ wurde gefunden[15] sowie eine mögliche, besondere Rolle des Immunproteasoms für den NF-κB Signalweg. Diese Funktionen werden jedoch in der Primärliteratur kontrovers diskutiert und sind daher noch nicht abschließend aufgeklärt[16].[17] In vielen Zellen des Immunsystems selbst wird dauerhaft das Immunproteasom neben dem Standardproteasom gebildet. Eine besonders große Menge an Immunproteasom findet sich in T- und B-Lymphozyten, die nahezu ausschließlich Immunproteasome beinhalten[18].
Thymoproteasom
Neben dem konstitutiven Proteasom und dem Immunproteasom gibt es eine dritte Klasse des Proteasoms, die ausschließlich in den kortikalen Epithelzellen des Thymus vorkommt. Hierbei wird die Untereinheit β5 durch die alternative Untereinheit β5t ("t" für thymoproteasome) ersetzt. Die kortikalen Epithelzellen des Thymus spielen eine wichtige Rolle beim Prozess der positiven Selektion während der Entwicklung der T-Lymphozyten. Fehlen alle alternativen Untereinheiten des 20S Proteasoms (β5i, β2i, β1i und β5t) in Mäusen, zeigen die Mäuse einen starken Defekt in der Reifung von T-Zellen, da diese zum größten Teil während der negativen Selektion verlorengehen. Diese Experimente lieferten Belege für das sogenannte Peptide Switching-Modell. Es besagt, dass die kortikalen Thymusepithelzellen durch Thymo- und Immunproteasom andere Peptide generieren als die medullären Thymusepithelzellen und die dendritischen Zellen bei der negativen Selektion, damit eine erfolgreiche Reifung der CD8-positiven T-Zellen erfolgen kann[19].
Zielproteine für den Abbau durch das Proteasom
Ein zentraler Prozess für den regulierten Abbau von Proteinen ist die sogenannte Ubiquitinierung. Proteine, die abgebaut werden sollen, werden in einem mehrstufigen enzymatischen Prozess von Ubiquitin-Protein-Ligasen mit einer Polyubiquitin-Kette markiert, welche von den 19S Komplexen erkannt wird. Ubiquitin ist ein kleines Protein mit einer Molekülmasse von 8,5 kDa. Die Polyubiquitin-Kette wird vor dem Einschleusen des Substrats ins 20S Proteasom in ihre einzelnen Ubiquitin-Moleküle zerlegt, die dann wiederverwendet werden können. Neben Ubiquitin kann die Markierung zum unmittelbaren Abbau durch das Proteasom auch durch den Ubiquitin-ähnlichen Modifikator FAT10 erfolgen. Im Gegensatz zu Ubiquitin wird FAT10 dabei jedoch mit dem Substrat abgebaut[20]. Bei Vorliegen von zellulärem Stress (z. B. Hitzeschock oder oxidativem Stress) werden vermehrt Proteine geschädigt, die reguliert abgebaut werden müssen. Oxidativ geschädigte Proteine und Proteine mit einer intrinsisch geringen Faltungsstabilität können teilweise auch von freien 20S Proteasomen direkt abgebaut werden[21].
Bedeutung des Proteasoms für die Zelle
Der kontrollierte Proteinabbau ist für die Zelle lebensnotwendig. So werden metabolische Enzyme, Transkriptionsfaktoren oder auch den Zellzyklus regulierende Proteine wie Cycline und CDK-Inhibitoren degradiert. Ebenso werden fehlerhafte Proteine abgebaut. Proteine werden in Abhängigkeit von ihrer Halbwertzeit reguliert abgebaut und bei Bedarf neu gebildet. Einige der vom Proteasom gebildeten Peptide werden ins Endoplasmatische Retikulum eingeschleust und anschließend an den Haupthistokompatibilitätskomplex I gebunden auf der Oberfläche der Zelle dem Immunsystem präsentiert (vgl. auch Abschnitt Immunproteasom). Ein Großteil der gebildeten Peptide wird jedoch im Zytosol durch weitere Peptidasen rasch in einzelne Aminosäuren zerlegt. Die Aminosäuren stehen dann für die Synthese neuer Proteine zur Verfügung. Im Gegensatz zu Kohlenhydraten und Fetten können Aminosäuren nicht in gesonderten Speicherpolymeren gespeichert werden. Aminosäuren müssen daher bei erhöhtem Proteinsynthese-Bedarf metabolisch gebildet werden, aus der Zellumgebung aufgenommen werden oder durch regulierten Abbau anderer zellulärer Proteine gewonnen werden. Daher sind Proteinsynthese und Proteinabbau durch intrazelluläre Signalprozesse eng miteinander verknüpft.[22] Eine nicht-ausreichende Fähigkeit von Zellen, beschädigte Proteine abzubauen, wurde in Verbindung mit neurodegenerativen Erkrankungen gebracht, in denen nicht abgebaute Proteinaggregate innerhalb und außerhalb von Zellen Schäden im zentralen und/oder peripheren Nervensystem verursachen. Ein Beispiel ist die Erkrankung Amyotrophe Lateralsklerose (ALS), die in Zusammenhang mit genetischen Veränderungen steht, welche u. a. eine Aggregation von Proteinen in Nervenzellen verursachen können. Durch Kryo-Elektronenmikroskopie wurden Hinweise gefunden, dass die resultierenden Proteinaggregate im Fall der C9orf72-Mutation (einer genetischen Variante, die mit ALS im Zusammenhang steht) eine Blockade des 26S Proteasoms verursachen, wodurch die normale Proteinqualitätskontrolle gestört wird.[23]
Proteasominhibitoren
Proteasominhibitoren sind chemische Substanzen, die die katalytische Aktivität des 20S Proteasoms oder das 26S Proteasom über den 19S Regulator hemmen[24]. Zu den schon früh entwickelten Proteasominhibitoren gehörten Peptidaldehyde wie der Inhibitor MG-132, der noch heute zu den am häufigsten verwendeten Proteasominhibitoren für die Erforschung proteasomabhängiger Prozesse in Zellen verwendet wird[25][26][27]. Anhaltende Inhibition des Proteasoms ist für Zellen toxisch. Wird das Proteasom am Abbau von Proteinen gehindert, resultiert daraus u. a. ein Mangel an notwendigen Aminosäuren in der Zelle. Dies ist einer der Mechanismen, durch den die Zellen in den Zelltod eintreten[28]. Proteasominhibitoren können zudem bestimmte Signalwege wie z. B. den NF-κB Signalweg blockieren. Daher können bestimmte anti-apoptotische Faktoren nicht mehr gebildet werden und die Zelle wird auch auf diesem Wege in den Zelltod geleitet[29][30].
Proteasominhibitoren in der klinischen Anwendung und präklinischen Entwicklung
Proteasominhibitoren finden in der klinischen Behandlung von Multiplem Myelom und Mantelzelllymphom Anwendung. Der erste, im Jahr 2003 klinisch zugelassene Proteasominhibitor war Bortezomib (Handelsname Velcade) und die Behandlungsoptionen wurden in jüngerer Zeit durch Proteasominhibitoren der zweiten Generation wie Carfilzomib und Ixazomib ergänzt. Aufgrund der Nebenwirkungen von Breitspektrum-Proteasominhibitoren wie Bortezomib ist der klinische Einsatz von Proteasominhibitoren zurzeit auf maligne Erkrankungen beschränkt. Zudem zeigen sich in der klinischen Anwendung Einschränkungen durch Resistenzentwicklung[31]. Zahlreiche weitere Proteasominhibitoren, deren Wirkung auf bestimmte Untereinheiten der verschiedenen Proteasome abzielt, werden derzeit präklinisch entwickelt und erforscht[32]. Dazu gehören auch selektive Immunproteasom-Inhibitoren wie die Wirkstoffe ONX 0914 oder KZR-616. Da diese Wirkstoffe gezielter die Untereinheiten des Immunproteasoms hemmen, besteht die Hoffnung, dass sie in Zukunft auch bei nicht-malignen Erkrankungen zum Einsatz kommen können wie z. B. bei Autoimmunerkrankungen[33][34] oder zur Behandlung von entzündlichen Erkrankungen wie der viralen Myokarditis[35].
Literatur
- Pollard T.D., Earnshaw W.C., Lippincott-Schwartz J. und G.T. Johnson: Cell Biology. 3. Auflage. Elsevier, 2017, ISBN 978-0-323-34126-4.
- Collins G. A. und A. L. Goldberg: The Logic of the 26S Proteasome. In: Cell. Band 169, Nr. 5, 2017, S. 792–806, doi:10.1016/j.cell.2017.04.023, PMID 28525752.
- A. Hershko: The ubiquitin system for protein degradation and someof its roles in the control of the cell division cycle. In: Cell Death and Differentiation. Band 12, 2005, S. 1191–1197, PMID 16094395.
- Niewerth D., G. Jansen, Y.G. Assaraf, S. Zweegman, G.J. Kaspers und J. Cloos.: Molecular basis of resistance to proteasome inhibitors in hematological malignancies. In: Drug Resistance Updates. Nr. 18, 2015, S. 18–35, doi:10.1016/j.drup.2014.12.001, PMID 25670156.
- Murata S., Y. Takahama, M. Kasahara und K. Tanaka: The immunoproteasome and thymoproteasome: functions, evolution and human disease. In: Nature Immunology. Band 19, Nr. 9, September 2018, S. 923–931, doi:10.1038/s41590-018-0186-z, PMID 30104634.
- Beling A. und M. Kespohl: Proteasomal Protein Degradation: Adaptation of Cellular Proteolysis With Impact on Virus—and Cytokine-Mediated Damage of Heart Tissue During Myocarditis. In: Frontiers in Immunology. Band 9, Nr. 2620, November 2018, S. eCollection 2018, doi:10.3389/fimmu.2018.02620.
- Basler M., S. Mundt, A. Bitzer, C. Schmidt und M. Groettrup: The immunoproteasome: a novel drug target for autoimmune diseases. In: Clinical and Experimental Rheumatology. Band 33, 4 Suppl 92, Juli 2014, S. S74–9, PMID 26458097.
- Budenholzer L., Cheng C.L., Li Y. und M. Hochstrasser: Proteasome Structure and Assembly. In: Journal of Molecular Biology. Band 429, Nr. 22, November 2017, S. 3500–3524, PMID 28583440.
- Limanaqi F., Biagioni F., Gaglione A., Busceti C.L. und F, Fornai: A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. In: Frontiers in Immunology. Band 10, Nr. 28, März 2019, S. eCollection 2019, PMID 30984192.
Einzelnachweise
- Stadtmueller, B. M. and C. P. Hill: Proteasome Activators. In: Molecular Cell. Band 41, Nr. 1, 2011, S. 8–19, doi:10.1016/j.molcel.2010.12.020, PMID 21211719.
- Whitby F. G., E. I. Masters, L. Kramer, J. R. Knowlton, Y. Yao, C. C. Wang and C. P. Hill: Structural basis for the activation of 20S proteasomes by 11S regulators. In: Nature. Band 408, Nr. 6808, S. 115–120, doi:10.1038/35040607, PMID 11081519.
- Osmulski P. A., M. Hochstrasser and M. Gaczynska: A Tetrahedral Transition State at the Active Sites of the 20S Proteasome Is Coupled to Opening of the α-Ring Channel. In: Structure. Band 17, Nr. 8, 2009, S. 1137–1147, doi:10.1016/j.str.2009.06.011, PMID 19679091.
- Wehmer M., T. Rudack, F. Beck, A. Aufderheide, G. Pfeifer, J. M. Plitzko, F. Förster, K. Schulten, W. Baumeister and E. Sakata: Structural insights into the functional cycle of the ATPase module of the 26S proteasome. In: Proceedings of the National Academy of Sciences of the United States of America. Band 114, Nr. 6, Februar 2017, S. 1305–1310, doi:10.1073/pnas.1621129114, PMID 28115689.
- Groll M., L. Ditzel, J. Löwe, D. Stock, M. Bochtler, H. D. Bartunik and R. Huber: Structure of 20S proteasome from yeast at 2.4Å resolution. In: Nature. Band 386, Nr. 6624, April 1997, S. 463-71, doi:10.1038/386463a0, PMID 9087403.
- Collins G. A. and A. L. Goldberg: The Logic of the 26S Proteasome. In: Cell. Band 169, Nr. 5, 2017, S. 792–806, doi:10.1016/j.cell.2017.04.023, PMID 28525752.
- Lasker K., F. Förster, S. Bohn, T. Walzthoeni, E. Villa, P. Unverdorben, F. Beck, R. Aebersold, A. Sali and W. Baumeister: Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. In: Proceedings of the National Academy of Sciences of the United States of America. Band 109, Nr. 5, Januar 2012, S. 1380-7, doi:10.1073/pnas.1120559109, PMID 22307589.
- Lander G. C., E. Estrin, M. E. Matyskiela, C. Bashore, E. Nogales and A. Martin: Complete subunit architecture of the proteasome regulatory particle. In: Nature. Band 482, Nr. 7384, Februar 2012, S. 186–191, doi:10.1038/nature10774, PMID 22237024.
- Orlowski, N., and S. Wilk: A multicatalytical protease complex from pituitary that forms enkephalin and enkephalin containing peptides. In: Biochemical and Biophysical Research Communications. Band 101, Nr. 3, August 1981, S. 814-22, PMID 7030330.
- Arrigo, A.-P., K. Tanaka, A. L. Goldberg, and W. J. Welch: Identity of the 19S “prosome” particle with the large multifunctional protease complex of mammalian cells (the proteasome). In: Nature. Band 331, Nr. 6152, Januar 1988, S. 192–194, doi:10.1038/331192a0, PMID 3277060.
- Nandi D., H. Jiang, J.J. Monaco: Identification of MECL-1 (LMP-10) as the Third IFN-γ-lnducible Proteasome Subunit. In: Journal of Immunology. Band 156, Nr. 7, April 1996, S. 2361-4, PMID 8786291.
- Groettrup, M., R. Kraft, S. Kostka, S. Standera, R. Stohwasser, and P. M. Kloetzel: A third interferon-gamma-induced subunit exchange in the 20S proteasome. Band 26, Nr. 4, April 1996, S. 863-9, doi:10.1002/eji.1830260421, PMID 8625980.
- Murata, S., K. Sasaki, T. Kishimoto, S.-I. Niwa, H. Hayashi, Y. Takahama, and K. Tanaka: Regulation of CD8+ T cell development by thymus-specific proteasomes. In: Science. Band 316, Nr. 5829, Juni 2007, S. 1349-53, doi:10.1126/science.1141915, PMID 17540904.
- Groettrup M., C. J. Kirk and M. Basler: Proteasomes in immune cells: more than peptide producers? In: Nature Reviews Immunology. Band 10, Nr. 1, Januar 2010, ISSN 1474-1741, S. 73–78, doi:10.1038/nri2687, PMID 20010787.
- Seifert U., Bialy L.P., Ebstein F., Bech-Otschir D., Voigt A., Schröter F., Prozorovski T., Lange N., Steffen J., Rieger M., Kuckelkorn U., Aktas O., Kloetzel P.M. and E. Krüger: Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. In: Cell. Band 142, Nr. 4, August 2010, S. 613-24, doi:10.1016/j.cell.2010.07.036, PMID 20723761.
- Beling A. and M. Kespohl: Proteasomal Protein Degradation: Adaptation of Cellular Proteolysis With Impact on Virus– and Cytokine-Mediated Damage of Heart Tissue During Myocarditis. In: Frontiers in Immunology. Band 9, Nr. 2620, November 2018, S. eCollection 2018, doi:10.3389/fimmu.2018.02620, PMID 30546359.
- Nathan J.A., Spinnenhirn V., Schmidtke G., Basler M., Groettrup M. and A.L. Goldberg: Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. In: Cell. Band 152, Nr. 5, Februar 2013, S. 1184-94, doi:10.1016/j.cell.2013.01.037, PMID 23452861.
- Schmidt C., T. Berger, M, Groettrup and M. Basler: Immunoproteasome Inhibition Impairs T and B Cell Activation by Restraining ERK Signaling and Proteostasis. In: Frontiers in Immunology. Band 9, Nr. 2386, Oktober 2018, S. eCollection 2018, doi:10.3389/fimmu.2018.02386, PMID 30416500.
- Kincaid E. Z., S. Murata, K. Tanaka, K. L. Rock: Specialized proteasome subunits have an essential role in the thymic selection of CD8(+) T cells. In: Nature Immunology. Band 17, Nr. 8, August 2016, S. 938–945, doi:10.1038/ni.3480, PMID 27294792, PMC 4955723 (freier Volltext).
- Aichem A., S. Anders, N. Catone, P. Rößler, S. Stotz, A. Berg, R. Schwab, S. Scheuermann, J. Bialas, M. C. Schütz-Stoffregen, G. Schmidtke, C. Peter, M. Groettrup and S. Wiesner: The structure of the ubiquitin-like modifier FAT10 reveals an alternative targeting mechanism for proteasomal degradation. In: Nature Communications. Band 9, Nr. 1, August 2018, doi:10.1038/s41467-018-05776-3, PMID 30127417.
- Ben-Nissan G. and M. Sharon: Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. In: Biomolecules. Band 4, Nr. 3, September 2014, S. 862–884, doi:10.3390/biom4030862, PMID 25250704.
- Zhang Y., J. Nicholatos, J.R. Dreier, S. J. H. Ricoult, S. B. Widenmaier, G. S. Hotamisligil, D. J. Kwiatkowski, B. D. Manning: Coordinated regulation of protein synthesis and degradation by mTORC1. In: Nature. Band 513, Nr. 7518, September 2014, S. 440-3, doi:10.1038/nature13492, PMID 25043031.
- Guo Q., C. Lehmer, A.Martínez-Sánchez, T. Rudack, F. Beck, H. Hartmann, M. Pérez-Berlanga, F. Frottin, M. S. Hipp, F. U. Hartl, D. Edbauer, W. Baumeister, R.Fernández-Busnadiego: In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. In: Cell. Band 172, Nr. 4, Februar 2018, S. 696–705.e12, doi:10.1016/j.cell.2017.12.030, PMID 29398115.
- Kisselev A. F., W. A. van der Linden, H. S. Overkleeft: Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. In: Chemistry & Biology. Band 19, Nr. 1, 2012, S. 99–115, doi:10.1016/j.chembiol.2012.01.003, PMID 22284358.
- Ito A., R. Takahashi, C. Muira, and Y. Baba: Synthetic Study of Peptide Aldehydes. In: Chemical and Pharmaceutical Bulletin. Band 23, Nr. 12, 1975, S. 3106–3113, doi:10.1248/cpb.23.3106.
- Satoshi Tsubuki, S., H. Kawasaki, Y. Saito, N. Miyashita, M. Inomata, and S. Kawashima: Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: A novel catalytic activity in proteasome. In: Biochem. Biophys. Res. Commun. Band 196, Nr. 3, November 1993, S. 1195-201, doi:10.1006/bbrc.1993.2378, PMID 8250877.
- Rock, K. L., C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang, and A. L. Goldberg: Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Band 78, Nr. 5, September 1994, S. 761–771, PMID 8087844.
- Suraweera A., C. Münch, A. Hanssum, A. Bertolotti: Failure of Amino Acid Homeostasis Causes Cell Death following Proteasome Inhibition. In: Molecular Cell. Band 48, Nr. 2, 2012, S. 242–253, doi:10.1016/j.molcel.2012.08.003, PMID 22959274.
- H. N. M. Ergin, Q. Huang, J. Qin, H. M. Amin, R. L. Martinez, S. Saeed, K.Barton, S. Alkan: Analysis of expression of nuclear factor κB (NF‐κB) in multiple myeloma: downregulation of NF‐κB induces apoptosis. In: British Journal of Haematology. Band 115, Nr. 2, Dezember 2001, S. 279-86, doi:10.1046/j.1365-2141.2001.03102.x, PMID 11703322.
- Ma M. H., H. H. Yang, K. Parker, S. Manyak, J. M. Friedman, C. Altamirano, Z. Wu, M. J. Borad, M. Frantzen, E. Roussos, J. Neeser, A.Mikail, J. Adams, N. Sjak-Shie, R. A. Vescio and J. R. Berenson: The Proteasome Inhibitor PS-341 Markedly Enhances Sensitivity of Multiple Myeloma Tumor Cells to Chemotherapeutic Agents. In: Clinical Cancer Research. Band 9, Nr. 3, März 2003, S. 1136-44, PMID 12631619.
- Niewerth D., G. Jansen, Y.G. Assaraf, S. Zweegman, G.J. Kaspers and J. Cloos.: Molecular basis of resistance to proteasome inhibitors in hematological malignancies. In: Drug Resistance Updates. Nr. 18, 2015, S. 18–35, doi:10.1016/j.drup.2014.12.001, PMID 25670156.
- Cromm P. M. and C. M. Crews: The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. In: ACS Central Science. Band 3, Nr. 8, 2017, S. 830–838, doi:10.1021/acscentsci.7b00252, PMID 28852696.
- Basler M., M. M. Lindstrom, J. J. LaStant, J. M. Bradshaw, T. D. Owens, C. Schmidt, E. Maurits, C. Tsu, H. S. Overkleeft, C. J. Kirk, C. L. Langrish and M. Groettrup: Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. In: EMBO reports. Oktober 2018, S. e46512, doi:10.15252/embr.201846512, PMID 30279279.
- Johnson H. W. B., E. Lowe, J. L. Anderl, A. Fan, T. Muchamuel, S. Bowers, D. Moebius, C. Kirk and D. L. McMinn: A required immunoproteasome subunit inhibition profile for anti-inflammatory efficacy and clinical candidate KZR-616 ((2S,3R)-N-((S)-3-(cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). In: Journal of Medicinal Chemistry. Oktober 2018, doi:10.1021/acs.jmedchem.8b01201, PMID 30380863.
- Althof N., C. C. Goetzke, M. Kespohl, K. Voss, A. Heuser, S. Pinkert, Z. Kaya, K. Klingel and A Beling: The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. In: EMBO Molecular Medicine. Band 10, Nr. 2, Februar 2018, S. 200–218, doi:10.15252/emmm.201708089, PMID 29295868.