UV/VIS-Spektroskopie
Die UV/VIS-Spektroskopie ist ein spektroskopisches Verfahren der optischen Molekülspektroskopie, das elektromagnetische Wellen des ultravioletten (UV) und des sichtbaren (englisch visible, VIS) Lichts nutzt. Die Methode ist auch unter UV/VIS-Spektralphotometrie oder als Elektronenabsorptionsspektroskopie[1] bekannt. Im Alltag werden die verwendeten Geräte häufig ungenau als Photometer bezeichnet.
Die UV/VIS-Spektroskopie umfasst einen relativ kleinen Wellenlängenbereich des elektromagnetischen Spektrums von etwa 200 nm bis 800 nm.[2] Für Routineanwendungen in der organischen Chemie wird der folgende Bereich verwendet: : 180-800 nm, : 55.556-12.500 cm-1.[3] Die Energie dieser Strahlung ist in der Lage, die Valenzelektronen der Atome auf ein höheres Energieniveau zu heben.


Prinzip
UV/VIS-Spektroskopie beruht auf Messung der Extinktion von sichtbarem und ultraviolettem Licht durch eine Probe. Die spektrale, d. h. wellenlängenabhängige, Information kann entweder durch Selektion und Scannen der Wellenlänge des einfallenden Lichts vor der Probe (siehe unten, Zweistrahl-Spektrometer) oder durch Trennen der Wellenlängen des transmittierten Lichts nach der Probe (siehe unten, Diodenarray-Spektrometer) gewonnen werden. Das Verhältnis der spektralen Intensität des transmittierten und des einfallenden Lichts liefert das Transmissionspektrum. Der logarithmische Kehrwert der Transmission ergibt das Extinktionsspektrum.
Grundsätzlich liefert die Extinktion Informationen über die Absorption, Streuung, Beugung und Reflexion an und in der Probe. Häufig werden in der UV/VIS-Spektroskopie Erscheinungen der Strahlungsabsorption ausgewertet, da die Photonenenergie des sichtbaren und ultravioletten Lichts der Übergangsenergie der Zustände von äußeren Elektronen vieler Atome und Moleküle entspricht. Durch Absorption von Photonen im sichtbaren und ultravioletten Spektralbereich können Valenzelektronen (beispielsweise die der p- und d-Orbitale) angeregt werden, das heißt, in einen Zustand höherer Energie übergehen. Das Transmissions- oder Extinktionsspektrum erlaubt daher die Identifikation und quantitative Bestimmung von Analyten.
Um ein Elektron beispielsweise von einem besetzten (HOMO) auf ein unbesetztes, höheres Orbital (LUMO) anzuheben, muss die Energie des absorbierten Photons genau der Energiedifferenz zwischen den beiden Energieniveaus entsprechen. Über den Zusammenhang

kann die Wellenlänge des absorbierten Lichtes für die aufzuwendende Energie berechnet werden, wobei die Energie, das plancksche Wirkungsquantum, die Lichtgeschwindigkeit, die Frequenz und die Wellenlänge der elektromagnetischen Welle sind. Dieser Zusammenhang wird manchmal auch als Einstein-Bohr-Gleichung bezeichnet.[4] Näherungsweise lässt sich dieser Zusammenhang in Form einer zugeschnittenen Größengleichung vereinfacht darstellen:





Stoffe, die nur im UV-Bereich ( < 400 nm) absorbieren, erscheinen dem menschlichen Auge farblos. Einen Stoff nennt man dann farbig, wenn er Strahlung im Bereich des sichtbaren Spektrums absorbiert. Dies ist sowohl bei Verbindungen zu erwarten, die über niedrige Anregungsenergien verfügen (π-zu-π*-Übergänge, konjugierten π-Elektronensystemen wie beispielsweise bei den Polyenen), als auch bei anorganischen Ionenkomplexen mit unvollständig aufgefüllten Elektronenniveaus (Beispiel: Cu2+-Verbindungen (meist bläulich – grünlich) gegenüber farblosen Cu+-Verbindungen). Ebenfalls erscheinen Verbindungen farbig, wenn stark polarisierende Wechselwirkungen zwischen benachbarten Teilchen bestehen, wie es z. B. beim gelben AgI der Fall ist. Bei nur einem Absorptionsgebiet nimmt das Auge die zur absorbierten Strahlung komplementäre Farbe wahr.[5]

Die Stärke der UV/VIS-Absorption einer Substanz ist für die Interpretation des Spektrums relevant. Die Extinktion (Absorbanz des Materials für Licht der Wellenlänge ) ist gegeben durch das Lambert-Beer’sche Gesetz:


ist die Schichtdicke des durchstrahlten Körpers (Einheit: m). Hierbei ist der dekadischer Extinktionskoeffizient (oft auch als spektraler Absorptionskoeffizient bezeichnet), welcher eine wellenlängenabhängige Stoffkonstante ist. Er wird in der Einheit bzw. angegeben. Als Ordinatengröße für UV/VIS-Spektren wird bzw. , wenn sich der -Wert im Spektrum über mehrere Zehnerpotenzen ändert, häufig verwendet.[2]





Anwendungen
_Complex.png.webp)
Die UV/VIS-Spektroskopie wird in der analytischen Chemie routinemäßig zur quantitativen Bestimmung verschiedener Analyten oder Proben eingesetzt, z. B. von Übergangsmetallionen, stark konjugierten organischen Verbindungen und biologischen Makromolekülen. Spektroskopische Analysen werden in der Regel in Lösungen durchgeführt, aber auch Feststoffe und Gase können untersucht werden.
Quantitative Bestimmung
Alle quantitativen Bestimmungen beruhen auf der Anwendung des Lambert-Beer'schen Gesetzes. Quantitative Bestimmungen werden durchgeführt bei[6]
- Konzentrationsbestimmungen,
- Bestimmung des molaren Extinktionskoeffizienten und
- reaktionskinetischen Messungen.
Qualitative Bestimmungen
UV/VIS-Spektren dienen dazu, Vermutungen über die Struktur der gesuchten Substanz zu bestätigen oder einzuschränken. Das Spektrum gibt Auskunft über die Art des Chromophors. Aussagen über andere Strukturteile des Moleküls sind nicht möglich. Die Bestimmung von Chromophoren mit Hilfe eines UV/VIS-Spektrums ist deshalb oft schwierig und wird daher in Kombination mit zusätzlichen spektroskopischen Methoden (IR-Spektroskopie, NMR-Spektroskopie) durchgeführt.[6]
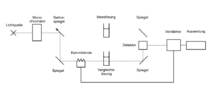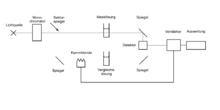
Aufbau eines Zweistrahl-UV/Vis-Spektrometers

Die Lichtquelle strahlt ultraviolettes, sichtbares und nahinfrarotes Licht im Wellenlängenbereich von etwa 200 nm bis 1100 nm aus. Im Monochromator wird zunächst die zur Messung ausgewählte Wellenlänge selektiert, worauf der Lichtstrahl auf den Sektorspiegel fällt. Der Sektorspiegel lässt das Licht abwechselnd durch die Messlösung und durch die Vergleichslösung fallen. Beide Lösungen befinden sich in sogenannten Küvetten. Die zwei Lichtstrahlen werden im Detektor empfangen und die Intensitäten im Verstärker verglichen. Der Verstärker passt dann die Intensität des Lichtstrahls aus der Vergleichslösung durch Einfahren der Kammblende der Intensität des Lichtstrahls aus der Messlösung an. Diese Bewegung wird auf einen Schreiber übertragen oder die Messwerte an eine Datenverarbeitung weitergegeben.
Zunehmend werden küvettenfreie UV/VIS-Spektrometer zur Konzentrationsbestimmung kleiner Probevolumina (unter 2 Mikroliter) von Proben höherer Konzentrationen eingesetzt.[7][8][9] Sogenannte Nanophotometer arbeiten mit Schichtdicken in Bereichen von 0,04 mm bis 2 mm. Sie benötigen keine Küvetten, keine Verdünnungen und können Proben mit einem Volumen von nur 0,3 µl analysieren (bei kleinster Schichtdicke), besitzen jedoch aufgrund der geringen Schichtdicke eine höhere Nachweisgrenze. Eine bewährte Technik basiert auf einer Kompression der Probe, welche somit unabhängig von der Oberflächenspannung und Verdunstung der Probe ist. Diese Methode findet Verwendung bei der Analyse von Nukleinsäuren (DNA, RNA, Oligonucleotide) und Proteinen (UV-Absorption bei 280 nm). Nach dem Lambert-Beer’schen Gesetz besteht ein Zusammenhang zwischen Absorption und Schichtdicke. Die Absorptionswerte bei den verschiedenen Schichtdicken (0,04 mm bis 2 mm) können somit berechnet werden. Geringe Schichtdicken wirken wie eine virtuelle Verdünnung der Probe, können jedoch nur bei entsprechend höheren Konzentrationen eingesetzt werden. Oftmals kann daher auf eine manuelle Verdünnung der Probe ganz verzichtet werden.
Aufbau eines Diodenarray-UV/VIS-Spektrometers


Eine weitere Technologie ist die Diodenarray-Technologie[10]. Die Probe in der Küvette wird mit einem Lichtstrahl bestrahlt, bestehend aus dem kontinuierlichen Wellenlängenbereich der Lichtquelle (z. B. Xenonblitzlichtlampe, 190 nm bis 1100 nm). Die Probe absorbiert bei einer Messung unterschiedliche Wellenlängen der Lichtquelle. Nicht absorbiertes Licht gelangt durch den Eintrittsspalt und wird an einem Beugungsgitter nach seiner Wellenlänge aufgespalten. Das Spektrum wird mithilfe eines CCD-Sensors detektiert und anschließend ausgewertet. Bei nicht automatisierten Geräten muss die Referenzprobe zusätzlich gemessen werden. Vorteile der Technologie sind kurze Messzeiten, da das gesamte UV/VIS-Spektrum mit einer Messung aufgenommen werden kann, ein niedriger Wartungsaufwand, da keine beweglichen Bauteile im Spektrometer vorhanden sind und dass die Geräte kompakt konstruiert werden können.
Theorie & chemische Beispiele
Bei der UV/VIS-Spektroskopie handelt es sich um eine Elektronenspektroskopie, das heißt, es werden Elektronen angeregt, genauer Valenzelektronen. Die Elektronenübergänge, die dabei auftreten können, lassen sich mit Hilfe der beteiligten Molekülorbitale bestimmen. Nach dem MO-Schema spalten sich die Orbitale bei Überlappung in besetzte bindende (σ, π) und nicht bindende Orbitale (n), sowie in nichtbesetzte antibindende Orbitale (π*, σ*) auf. In der Regel befinden sich alle Elektronen im bindenden Orbital und das antibindende Orbital bleibt leer. Werden Elektronen angeregt, wechseln sie in das antibindende Orbital. Man unterscheidet daher folgende Elektronenübergänge, die mit σ→σ*, π→π*, n→π* usw. bezeichnet werden. σ-Orbitale liefern eine wesentlich größere Aufspaltung als π-Orbitale, da die Energieaufspaltung zwischen dem bindenden und dem antibindenden Orbital mit dem Grad der Überlappung der Atomorbitale zunimmt.[11]

Die für die Absorption verantwortlichen Molekülbestandteile werden als Chromophore (griechisch: farbtragend) bezeichnet.[12]

Elektronenabsorption organischer Verbindungen
Organische Verbindungen, insbesondere solche mit hohem Konjugationsgrad, absorbieren auch Licht im UV- oder sichtbaren Bereich des elektromagnetischen Spektrums.
Verbindungen mit einem σ→σ*-Übergang sind z. B. Alkane wie Hexan, Cyclohexan und Heptan; mit einem n→σ*-Übergang sind z. B. aliphatische Verbindungen mit Heteroatomen (Alkohole); mit einem π→π*-Übergang sind z. B. Alkene und Alkine; mit einem n→π*-Übergang sind z. B. Verbindungen mit mesomeren Strukturelementen (Carbonylgruppe, Nitrogruppe, Nitrilgruppe). Die π-zu-π*-Übergänge erfolgen über längerwelliges UV-Licht und sind einfach zu messen. Man erhält Informationen über die absorbierende Wellenlänge des Moleküls, die Struktur und Farbe. Dabei erfolgt die Lichtabsorption im umso längerwelligen Bereich, je größer die Anzahl der konjugierten Doppelbindungen ist.[13] Mit Hilfe der Woodward-Fieser-Regel kann die Lage des Absorptionsmaximums des π→π*-Übergangs ( in nm) abgeschätzt werden. Liegt die Energie der π-zu-π*-Übergänge im Bereich des sichtbaren Lichts, so erscheint das Molekül farbig. Dabei nimmt es immer die Komplementärfarbe des absorbierten Lichts an. Beispiele für UV/VIS-absorbierende Chemikalien sind in der nachstehenden Tabelle aufgeführt:

| Beispiel | Chromophor | Übergang | / nm | / |
|---|---|---|---|---|
| σ → σ* | 122 | intensiv | ||
| σ → σ* | 135 | intensiv | ||
| n → σ* | 177 | 200 | ||
| n → σ* | 193 | 2500 | ||
| π → π* | 162 | 15000 | ||
| π → π* | 197 | 11000 | ||
| n → π* | 279 | 15 | ||
| n → π* | 200 | 50 |















Die für diese UV/VIS-Messungen verwendeten Lösungsmittel sind häufig Wasser für wasserlösliche Verbindungen oder Ethanol für organisch lösliche Verbindungen. Nur bei Messungen im gasförmigen Zustand gibt es keinen Einfluss durch die Umgebung, aber in gelöster Form (kondensierte Phase) sind veränderte Spektren aufgrund von Lösungsmitteleffekten sichtbar. Organische Lösungsmittel können eine beträchtliche UV-Absorption aufweisen. Daher sind nicht alle Lösungsmittel für die UV/VIS-Spektroskopie geeignet. Ethanol absorbiert bei den meisten Wellenlängen sehr schwach. Die Polarität des Lösungsmittels und der pH-Wert können das Absorptionsspektrum einer organischen Verbindung beeinflussen.[1] So zeigt der Reichardt-Farbstoff in Wasser ein Absorptionsmaximum bei 453 nm, in Ethanol bei 550 nm und in Diphenylether bei 810 nm.[3]
Elektronenabsorption anorganischer Verbindungen
Auch anorganische Verbindungen zeigen oft starke Farben, d. h. sie absorbieren im UV/VIS-Bereich. Bei kleinen anorganischen Molekülen wird, ähnlich wie bei organischen Molekülen, die MO-Theorie zur Erklärung herangezogen.[11][15]
- Wasser: farblos, da n→σ*-Übergang ()
- Ammoniak: farblos, da n→σ*-Übergang ()
- Stickstoffdioxid: braun, da n→π*-Übergang ()



Übergangsmetalle als Zentralion mit anorganischen / organischen Liganden bilden intensiv gefärbte Komplexe. Die Ligandenfeldtheorie liefert einen Erklärungsansatz für die Farbigkeit vieler Komplex- bzw. Koordinationsverbindungen. In der Ligandenfeldtheorie werden die Wechselwirkungen zwischen den Liganden des Komplexes und den d-Elektronen des Zentralions berücksichtigt. Diese besagt, dass es je nach d-Orbital und räumlicher Anordnung (z. B. oktaedrisch, tetraedrisch) sowie Anzahl und Art der Liganden energetisch günstigere und ungünstigere d-Orbitale gibt, weshalb die d-Orbitale in zwei Gruppen eingeteilt werden. Durch die energetische Aufspaltung der d-Orbitale im Ligandenfeld kann ein Elektron aus einem besetzten, tiefer gelegenen d-Orbital in ein energetisch höher gelegenes d-Orbital angehoben werden. Dadurch werden in Übergangsmetallkomplexen d-d-Übergänge zwischen den d-Orbitalen im Ligandenfeld möglich. Weiterhin treten Übergänge innerhalb des Liganden auf, bei denen häufig π→π*-Übergänge auftreten. Neben diesen Übergängen in Koordinationsverbindungen sind auch Charge-Transfer-(CT-)Übergänge möglich. Hierbei wird ein Elektron aus einem (ungeraden) p-Orbital in ein (gerades) d-Orbital angeregt. Vier dieser Übergänge sind möglich: Ligand-zu-Metall-CT-Übergänge (LMCT) und Elektronenübergänge von besetzten d-Orbitalen zu leeren Ligandenorbitalen (Metall-zu-Ligand-CT-Übergang, MLCT), sowie Metall-zu-Metall-CT-Übergänge (MMCT) und Ligand-zu-Ligand-CT-Übergänge (LLCT, kommen nicht häufig vor).[16]
Folglich sind Übergangsmetallkomplexe durch drei Arten von Absorptionen gekennzeichnet:[17]
- Innerligand-Übergänge ()
- CT-Übergänge ()
- d-d-Übergänge ()



Regeln zu Elektronenübergängen
Bei den betrachteten Elektronenübergängen sind stets die folgenden Auswahlregeln zu beachten (u. a. Laporte-Regel):
- Spinregel: Der Gesamtspin muss erhalten bleiben (, wobei mit sind die Spinmomente der einzelnen Elektronen), sonst spinverboten.[11][2]
- Bahnregel: Die Differenz der Gesamt-Bahndrehimpulse darf nur die Werte (wobei mit sind die Bahnmomente der einzelnen Elektronenannehmen), sonst bahnverboten.
- Übergänge zwischen verschiedenen Spinmultiplizitäten () sind verboten.[11] Eine Anregung ist nur erlaubt, wenn sich der Gesamtspin des Moleküls nicht ändert, also wenn vor und nach der Anregung die gleiche Anzahl an gepaarten und ungepaarten Elektronen (Spins) vorhanden ist.
- Laporte-Verbot: Das Verbot bezieht sich auf Symmetrieänderungen bei Elektronensprüngen.[2] Ein gerader Zustand nach Laporte liegt vor, wenn in Mehrelektronensystemen die Summe der Bahndrehimpulsquantenzahlen gerade ist, andernfalls ist der Zustand ungerade. Verboten sind somit Sprünge, bei denen sich die Symmetrie nicht ändert bzw. die Parität gleich bleibt.







Bei dem Laporte-Verbot müssen nach der Ligandenfeldtheorie zwei Abfragen getätigt werden.
- Besitzt das Molekül ein Inversionszentrum (symmetrisch in Bezug auf die Punktspiegelung)? Wenn nein (z. B. Tetraeder), dann ist die Anregung zulässig; wenn ja (z. B. Oktaeder), dann ist eine Anregung erstmal nicht erlaubt und es muss Abfrage 2 beachtet werden.
- Ändert sich die Parität (das Vorzeichen) der Orbitale? Wenn ja, dann ist die Anregung erlaubt (also z. B. der Übergang von p → d-Orbital). Wenn nein, dann ist die Anregung nicht erlaubt (z. B. der Übergang von d → d-Orbital). Ein Übergang darf also nur von gerade nach ungerade, oder von ungerade nach gerade stattfinden (gerade sind die s- und d-Orbitale, ungerade die p- und f-Orbitale).
Kristallfeldübergänge, bei denen Elektronen von einem d- in ein d-Orbital angeregt werden, sind daher in oktaedrischen und anderen Komplexen mit Inversionszentrum verboten, in tetraedrischen und anderen Komplexen ohne Inversionszentrum jedoch erlaubt.[16][18] Weitere Beispiele für verbotene oder erlaubte Übergänge in einem oktaedrischen Komplex sind:
- verboten ist der Übergang 3s → 4s
- erlaubt ist der Übergang 3s → 4p
Diese vier Verbote bedeuten nicht, dass diese Übergänge nicht vorkommen, sondern nur, dass sie sehr unwahrscheinlich sind. Die schwache Farbe der Komplexe wird durch Schwingungen der Liganden relativ zum Metallzentrum verursacht. Dadurch wird die für das Laporte-Verbot wichtige Inversionssymmetrie kurzzeitig aufgehoben und ein Übergang kann stattfinden. Erlaubte Übergänge sind an -Werte von meist zu erkennen. Verbotene Übergänge sind im Spektrum an kleinen -Werten () zu erkennen.[2]


UV/VIS-Spektreninterpretation
Ein UV/VIS-Spektrum enthält keine scharfen Linien (wie das Linienspektrum eines Atoms), sondern breite Banden. Moleküle haben Bandenspektren. Banden bestehen aus vielen eng benachbarten Spektrallinien. Ein UV/VIS-Spektrum ist durch die Lage und Intensität der Absorptionsbande gekennzeichnet. Die Abszissengröße (x-Achse) des Spektrums ist meist die Wellenlänge in nm oder die Wellenzahl in cm-1, die beschreiben, in welchem Bereich des elektromagnetischen Spektrums absorbiert wird. Als Ordinatengröße (y-Achse) wird meist der dekadische Extinktionskoeffizient bzw. in M-1cm-1 verwendet. Dieser beschreibt die Stärke der Absorption.


In Kombination mit anderen spektroskopischen Untersuchungsmethoden (IR-Spektroskopie, NMR-Spektroskopie bspw. 1H-NMR) kann die UV/VIS-Spektroskopie eine dienliche Methode zur qualitativen Analyse und Strukturbestimmung sein.[11] Das UV/VIS-Spektrum gibt Auskunft über die Art des Chromophors.[6] Diese Spektren dienen somit lediglich dazu, Vermutungen über die Struktur der gesuchten Substanz zu bestätigen oder einzuschränken. Innerhalb einer „Stofffamilie“ treten oft typische Absorptionen auf, die bei Vorliegen bestimmter Strukturen eine einfache Identifizierung ermöglichen.[6] Es ist nicht möglich, aus einem UV/VIS-Spektrum ohne weitere Informationen auf die Molekülstruktur zu schließen.[6]
Eine wertvolle Hilfe bei der qualitativen Interpretation von Spektren sind die Strukturaufklärungstabellen für organische Verbindungen, die in verschiedenen Büchern zu finden sind.
| Intensität | / | Übergang | Chromophor |
|---|---|---|---|
| schwach | 10–100 | n → π* | in Ketonen (ca. 260 nm) |
| mittel | 1000–10000 | π → π* | Aromaten |
| stark | 10000–20000 | n → π*
π → π* |
einfach konjugierte Systeme:
Diene, α, β-ungesättigte Ketone, substituierte Aromaten |
| sehr stark | >20000 | n → π*
π → π* |
mehrfach konjugierte Systeme,
substituierte Aromate |
Siehe auch
Weblinks
Einzelnachweise
- Manfred Reichenbächer, Jürgen Popp: Strukturanalytik organischer und anorganischer Verbindungen: Ein Übungsbuch. Springer Science & Business Media, 2007, ISBN 978-3-8351-0190-6, S. 119 (eingeschränkte Vorschau in der Google-Buchsuche).
- Wolfgang Bechmann, Ilko Bald: Einstieg in die Physikalische Chemie für Naturwissenschaftler. doi:10.1007/978-3-662-55858-4 (springer.com [abgerufen am 13. April 2023]).
- Martin Badertscher: ANALYTISCHE CHEMIE für Biologen und Pharmazeuten. 2011, abgerufen am 18. April 2023.
- Christopher G. Morris, Academic Press: Academic Press dictionary of science and technology. Gulf Professional Publishing, 1992, ISBN 0-12-200400-0, S. 716 (eingeschränkte Vorschau in der Google-Buchsuche).
- Autorenkollektiv, federführende Autoren: K. Doernel, R. Geyer: Analytikum – Methoden der analytischen Chemie und ihre theoretischen Grundlagen. 8. Auflage. VEB Deutscher Verlag für Grundstoffindustrie, Leipzig 1990, ISBN 3-342-00191-7, S. 259 ff.
- P. Wörfel, M. Bitzer, U. Claus, H. Felber, M. Hübel, B. Vollenweider: Laborpraxis. Springer Basel AG, 1990, doi:10.1007/978-3-0348-6162-5 (springer.com [abgerufen am 14. April 2023]).
- H. Stranneheim, J. Lundeberg: Stepping stones in DNA sequencing. In: Biotechnology journal. Band 7, Nummer 9, September 2012, ISSN 1860-7314, S. 1063–1073, doi:10.1002/biot.201200153, PMID 22887891, PMC 3472021 (freier Volltext).
- P. O. Michel, C. Degen, M. Hubert, L. Baldi, D. L. Hacker, F. M. Wurm: A NanoDrop-based method for rapid determination of viability decline in suspension cultures of animal cells. In: Analytical biochemistry. Band 430, Nummer 2, November 2012, ISSN 1096-0309, S. 138–140, doi:10.1016/j.ab.2012.08.028, PMID 22960013.
- M. T. Kelliher, M. S. Piraino, M. E. Gemoules, C. A. Southern: A comparison of Förster resonance energy transfer analysis approaches for Nanodrop fluorometry. In: Analytical biochemistry. Band 441, Nummer 1, Oktober 2013, ISSN 1096-0309, S. 44–50, doi:10.1016/j.ab.2013.06.009, PMID 23811157.
- Tony Owen: Fundamentals of Modern UV-Visible Spectroscopy: A Primer. 1996, OCLC 918510068.
- Stefan Bienz, Laurent Bigler, Thomas Fox, Herbert Meier: Spektroskopische Methoden in der organischen Chemie. 9. Auflage. Georg Thieme Verlag, Stuttgart 2016, ISBN 978-3-13-576109-1, doi:10.1055/b-004-129729 (thieme.de [abgerufen am 13. April 2023]).
- Roland Winter, Frank Noll: Methoden der Biophysikalischen Chemie. Vieweg+Teubner Verlag Wiesbaden, 1998, doi:10.1007/978-3-663-05794-9 (springer.com [abgerufen am 14. April 2023]).
- Joseph B. Lambert, Scott Gronert, Herbert F. Shurvell, David A. Lightner: Spektroskopie – Strukturaufklärung in der Organischen Chemie 2. Auflage, Pearson Deutschland, München 2012, ISBN 978-3-86894-146-3, S. 591–653.
- Ernö Pretsch, Philippe Bühlmann, Martin Badertscher: Spektroskopische Daten zur Strukturaufklärung organischer Verbindungen. doi:10.1007/978-3-540-76866-1 (springer.com [abgerufen am 13. April 2023]).
- E Hawe, C Fitzpatrick, P Chambers, E Lewis: An investigation into the use of an integrating sphere as a gas absorption cell. In: Journal of Optics A: Pure and Applied Optics. Band 9, Nr. 6, 1. Juni 2007, ISSN 1464-4258, S. S12–S18, doi:10.1088/1464-4258/9/6/S03 (iop.org [abgerufen am 14. April 2023]).
- Birgit Weber: Koordinationschemie. doi:10.1007/978-3-662-63819-4 (springer.com [abgerufen am 13. April 2023]).
- Manfred Reichenbächer, Jürgen Popp: Strukturanalytik organischer und anorganischer Verbindungen. Vieweg+Teubner Verlag Wiesbaden, 2007, doi:10.1007/978-3-8351-9204-1 (springer.com [abgerufen am 14. April 2023]).
- Koordinationschemie. Ludwig-Maximilians-Universität München, abgerufen am 13. April 2023.