Steißbeinteratom
Das Steißbeinteratom oder Steißteratom (lat. teratoma sacrococcygeale), ein Teratom im Bereich des Steißbeins, ist die häufigste Manifestation von Keimzelltumoren des Kleinkindalters. Es bezeichnet einen Fehlbildungstumor im unteren Bereich der fetalen Wirbelsäule, der bis in das Becken des Ungeborenen vorwachsen kann.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| D48.0 | Neubildung unsicheren oder unbekannten Verhaltens an sonstigen und nicht näher bezeichneten Lokalisationen – Knochen und Gelenkknorpel |
| O33.7 | Betreuung der Mutter bei Missverhältnis durch sonstige Deformitäten des Fetus |
| ICD-10 online (WHO-Version 2019) | |
Pathologie
Ein Teratom der Mittellinie entspringt einer Blastulazelle (abortiver Zwilling)[2] Histologisch handelt es sich um lymphangiomatöse lipomatöse Wucherungen, die Abkömmlinge aller drei Keimschichten enthalten.[2]
Der Ursprung wird in ventral des Steißbeines gelegenen Zellen des Primitivstreifens (Hensenscher Knoten) vermutet mit Entwicklung um die 2.–3. Gestationswoche.
Meistens ist das Teratom gemischt solide-zystisch, rein zystisch in etwa 15 %[1]
Vorkommen und Häufigkeit
Die Häufigkeit von Steißteratomen wird mit 1 zu 40.000 Lebendgeborenen angegeben, das Verhältnis weiblich zu männlich liegt bei 4 zu 1.[2]
Einteilung
Eine Einteilung kann aufgrund pathologischer Kriterien erfolgen:[1][2]
- gutartig, reifes Teratom, häufigste Form, etwa 60–70 %
- unreifes Teratom
- embryonales Teratom
- Teratokarzinom in 10–30 %
Klassifikation
Auf der Lokalisation und Wachstumsrichtung basiert die Klassifikation der American Academy of Pediatric Surgery Section Survey:[3][2]
- Typ I: „Tiefe mediale Form“, Wachstum „extra-fetal“ hinter dem Rektum nach kaudal, häufigste Form, etwa 47 %
- Typ II: Wachstum „extra-fetal“ mit Ausdehnung vor dem Sakrum ins kleine Becken
- Typ III: Wachstum „extra-fetal“ mit Ausdehnung in Richtung Abdomen
- Typ IV: Wachstum ausschließlich innerhalb des Beckens
Klinische Erscheinung
Dieser Tumor kann, durch die falsche Entfaltung der Keimblätter, alle möglichen Arten von Gewebe bis hin zu Organen oder weiteren Gliedmaßen beinhalten. In über 90 % der Fälle sind Steißbeinteratome gutartig, neigen jedoch dazu, bösartig zu werden. Bis zur Geburt können sie bereits kindskopfgroß sein. Eine Untergruppe der Tumoren wird so stark durchblutet, dass hierdurch ein schweres fetales Herzversagen bis hin zum Versterben des Ungeborenen auftreten kann.
Diagnose
Für die Diagnose ist wesentlich, zu welchem Zeitpunkt das Teratom erkennbar wird:
- Fetal, bereits im Mutterleib, durch Ultraschall während einer Routineuntersuchung oder beim Feinultraschall
- Neonatal, beim Neugeborenen, zumeist klinisch
- erst im Kleinkindesalter mit höherer Wahrscheinlichkeit einer vorliegenden Malignität von 48 % – 67 % für Mädchen bzw. Jungen älter als 2 Monate
Im Blutserum kann das Alpha-1-Fetoprotein und das Beta-HCG erhöht sein.
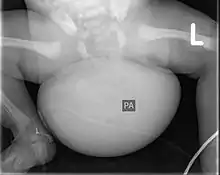

Zur Bildgebung kann der Tumor, dessen Struktur, Ausdehnung und Durchblutung gut im Ultraschall sowie in der Kernspintomographie dargestellt werden, im Röntgenbild können Kalk- und Knochenanteile sichtbar werden.[4]
Es besteht eine Assoziation mit Meningomyelozele und Wirbelkörperanomalien.[1]
Als Komplikationen können auftreten:[1][5]
- Obstruktion der Harnwege oder des Gastrointestinaltraktes
- Kompression von Nerven oder des Plexus lumbosacralis
- Anämie
- Shunts mit high output failure
- Geburtshindernis
- Tumorruptur während der Geburt
Differentialdiagnostik
Abzugrenzen sind:[2]
- tiefe Meningozele, Meningomyelocele mit Lipom
- terminale Enterogene Zyste
- Rektumduplikatur
- Sakrales Chordom
- Fibrosarkom
- Neurofibrom
- Ganglioneurom
- Ependymom
- Riesenzelltumor
Therapie
Vorgeburtlich kann mittels fetaler Chirurgie zwischen der 20. bis zur 32. Schwangerschaftswoche eine Verminderung der Tumordurchblutung und eine Verbesserung der Kreislaufsituation erreicht werden. Sofern keine vorgeburtliche Behandlung erfolgte, wird es frühstmöglich nach der Geburt operativ entfernt, einschließlich Teilen des Steißbeines.[2]
Prognose
Etwa 80 % sind gutartig.[1] Die Malignitätswahrscheinlichkeit steigt bei Manifestation nach dem Neugeborenenalter.[2]
Literatur
- W. Schuster, D. Färber (Herausgeber): Kinderradiologie. Bildgebende Diagnostik. Springer 1996, Band II, S. 399 f., ISBN 3-540-60224-0.
Einzelnachweise
- Radiopaedia
- Marcel Bettex (Hrsg.), Max Grob (Begr.), D. Berger (Bearb.), N. Genton, M. Stockmann: Kinderchirurgie. Diagnostik, Indikation, Therapie, Prognose. 2., neubearbeitete Auflage. Thieme, Stuttgart / New York 1982, S. 10.26ff, ISBN 3-13-338102-4
- R. P. Altman, J. G. Randolph, J. R. Lilly: Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. In: Journal of pediatric surgery. Band 9, Nummer 3, Juni 1974, S. 389–398, PMID 4843993.
- H. M. Yoon, S. J. Byeon, J. Y. Hwang, J. R. Kim, A. Y. Jung, J. S. Lee, H. K. Yoon, Y. A. Cho: Sacrococcygeal teratomas in newborns: a comprehensive review for the radiologists. In: Acta radiologica. [elektronische Veröffentlichung vor dem Druck] Januar 2017, doi:10.1177/0284185117710680, PMID 28530139.
- P. Kohlberger, A. Schaller: Das Steißteratom in der Geburtshilfe. In: Zeitschrift für Geburtshilfe und Neonatologie. Band 204, Nr. 3, 2000 May-Jun, S. 106–113, doi:10.1055/s-2000-10205, PMID 10909166.