Selenoketone
Selenoketone (Selone) sind organische, chemische Verbindungen. Sie stellen die Selen-Analoga von Ketonen dar und gehören zu den Selenocarbonylverbindungen.
| Selenoketone |
|---|
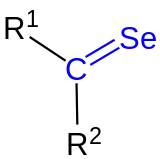 |
| Die Selenocarbonylgruppe ist blau markiert. Die Reste R1 und R2 sind organische Reste (Alkyl, Aryl o. ä.). |
Ihre funktionelle Gruppe besteht aus einem Kohlenstoff- und einem Selenatom, welche durch eine Doppelbindung miteinander verbunden sind. Bei den Resten R1 und R2 am Kohlenstoffatom der Selenocarbonylgruppe handelt es sich um organische Reste (Alkylrest, Arylrest etc.). Selenoketone mit gleichen Resten (R1 = R2) sind symmetrische Selenoketone, während Selenoketone mit ungleichen Resten (R1 ≠ R2) als usymmetrische Selenketone bezeichnet werden.[1] Analog zu den anderen Carbonylverbindungen der Chalkogengruppe (z. B. Thiocarbonylverbindungen) handelt es sich um eine reaktive Stoffklasse. Heute sind einige Methoden zur Herstellung stabiler Selenoketone bekannt. Sie können als chirale Derivatisierungsreagenzien zur stereoselektiven Kontrolle von Aldolreaktionen eingesetzt werden.[2] Außerdem eignen sie sich für die Einführung von Selenatomen in heterocyclische Verbindungen. Sowohl einfache als auch sterisch anspruchsvolle Selenoketone können als Dimere auftreten.[3] Selenoketone sind dafür bekannt, mit 1,3-Dienen Cycloadditionen in Analogie zur Diels-Alder-Reaktionen einzugehen.
Synthese
Die Synthese von Selenoketonen kann aus Ketonen, wie Aceton erfolgen, indem diese säurekatalysiert mit Selenwasserstoff zur Reaktion gebracht werden. Dabei entsteht Selenoaceton:[3]

Da Selenwasserstoff extrem toxisch ist, wurden weitere Synthesen entwickelt. Beispielsweise können Phosphor-Ylide mit elementarem Selen umgesetzt werden. So kann man z. B. Selenobenzophenon synthetisieren.[4] Alternativ können Hydrazone der Phosphor-Ylide mit Selen pyrolysiert werden. Diese Variante ermöglicht viele Synthesevarianten stabiler, sterisch stark gehinderter Selenoketone in guten Ausbeuten. Hydrazone können außerdem mit Selen(I)-chlorid oder -bromid und Triethylamin zu Selenoketonen umgesetzt werden.[5][6] Die Einwirkung von Bis(dimethylaluminium)selenid auf Ketone wird ebenfalls als geeignetes Verfahren beschrieben.[7] Selenoketone können außerdem durch die [3,3]-sigmatrope Umlagerung von Allyl-Alkenseleniden synthetisiert werden.[8]
Reaktionen
Obwohl die Synthese von stabilen, monomeren Selenoketonen möglich ist, werden die Produkte häufig als Dimere erhalten. Zwei Monomere des Selenoacetons dimerisieren z. B. zum Dimer Diselenoaceton.[3]

Dieses wird als klares, rotes Öl isoliert und riecht nach Knoblauch. Die Dimerisierung ist oft reversibel. Das Monomer von Selenobenzophenon ist stabil, kann aber in das Dimer überführt werden.[4] Instabile Monomere der Selenoketone können vinylogisch (Vinylogie-Prinzip), durch sterisch anspruchsvolle Reste (z. B. tert-Butylgruppen) oder als Liganden in Komplexen mit Übergangsmetallen (z. B. Nickel, Platin[9]) stabilisiert werden.[5][10] Die Ausbildung des Komplexes verringert die Reaktivität der Selenoketone und erhöht so ihre Handhabbarkeit.[11] Des Weiteren gelten Selenoketone als gute Dienophile in Diels-Alder-Reaktionen mit 1,3-Dienen. Dieses Verfahren ist gut geeignet, um Selen in Heterocyclen einzuführen. Durch die Umsetzung von Selenoaceton mit 1,3-Butadien wird das Selenopyran selektiv erhalten.[4]
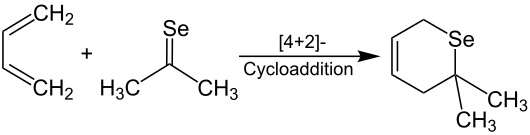
Zudem können Selenoketone mit Natriumborhydrid zu Selenolen reduziert werden. Die anschließende Oxidation an Luftsauerstoff führt zu Diseleniden.[1]
Einzelnachweise
- Paulmier, C. (1986): Selenium Reagents and Intermediates in Organic Synthesis. 4. Band. Oxford: Pergamon Books. S. 60–63, ISBN 0-08-032484-3.
- Silks, L. et al.: Chiral N-Acetyl Selone-Promoted Aldol Reactions. Synthetic Communications, 2009, 39(4), S. 641–653. doi:10.1080/00397910802419706.
- Schönberg, A. & Wagner, A. (1955): Methoden zur Herstellung und Umwandlung von Thioaldehyden und Thioketonen. In Müller, E. (Hrsg.): Methoden der Organischen Chemie. Band IX: Schwefel-, Selen-, Tellurverbindungen. Stuttgart: Thieme Verlag. S. 1199–1202.
- Erker, G. eta al.: Synthesis and Cycloadditions of Monomeric Selenobenzophenone. Angewandte Chemie International Edition, 1990, 29(9), S. 1067. doi:10.1002/anie.199010671.
- Guzirc, F.S. (1987): The Chemistry of Selencarbonylcompounds. In Liotta, D. (Ed.): Organoselenium Chemistry. New York: Wiley. S. 277–324, ISBN 0-471-88867-2.
- Ishii, A., Okazaki, R. & Inamoto, N.: Synthesis and reactions of selenoketones. Bulletin of the Chemical Society of Japan, 1988, 61(3), S. 861–867. doi:10.1246/bcsj.61.861.
- Segi, M. eta al.: Novel route to selenoketones from ketones by the use of bis (dimethylaluminum) selenide. Tetrahedron, 1989, 30(16), S. 2095–2098. doi:10.1016/S0040-4039(01)93721-9.
- Shimada, K. eta al.: Generation of a Selenoaldehyde, a Selenoketone, and Telluroaldehydes by [3, 3] Sigmatropic Rearrangement of Allyl Alkenyl Selenides and Tellurides. Chemistry letters, 1995, 24(2), S. 135–136. doi:10.1246/cl.1995.135.
- Shigetomi, T. et al.: Synthesis of Selenoketone–Platinum Complex. Bulletin of the Chemical Society of Japan, 2007, 80(2), S. 395–399. doi:10.1246/bcsj.80.395.
- Okazaki, R. & Tokitoh, N.: Heavy ketones, the heavier element congeners of a ketone. Accounts of Chemical Research, 2000, 33(9), S. 625–630. doi:10.1021/ar980073b. PMID 10995200.
- Fischer, H. et al.: [4+ 2]‐Cycloadditionen mit übergangsmetallkoordinierten Selenoaldehyden und Selenoketonen als Heterodienophile. Angewandte Chemie, 1986, 98(1), S. 80–81. doi:10.1002/ange.19860980109.