RESOLFT-Mikroskopie
Die RESOLFT-Mikroskopie (englisch reversible saturable optical linear (fluorescence) transitions, dt. ‚reversibel sättigbare optische (Fluoreszenz-) Übergänge‘) ist eine Gruppe von lichtmikroskopischen Verfahren, bei der man besonders scharfe Bilder erhält. Trotz Verwendung herkömmlicher Objektive und gebeugter Strahlen wird eine Auflösung weit jenseits der Beugungsgrenze bis herunter auf die molekulare Skala erhalten.
In einem herkömmlichen Lichtmikroskop können keine Details unterschieden werden, die enger beieinander liegen als circa 200 nm. Diese Einschränkung beruht auf der Wellennatur des Lichts. In herkömmlichen Lichtmikroskopen ist diese Auflösungsgrenze im Wesentlichen durch die Wellenlänge des verwendeten Lichts und die numerische Apertur bestimmt. Die RESOLFT-Mikroskopie überwindet diese Grenze, indem sie die Farbstoffe vorübergehend in einen Zustand schaltet, in dem sie nicht in der Lage sind, nach Beleuchtung mit einem (Fluoreszenz-)Signal zu antworten.
Funktionsprinzip

Die RESOLFT-Mikroskopie ist eine Variante der Lichtmikroskopie. Sie überwindet die Beugungsgrenze, indem sie die Details eines Präparats, die normalerweise zu dicht beieinander liegen, um aufgelöst zu werden, nacheinander aufnimmt. Damit wird das Prinzip der STED-[1][2] und GSD-Mikroskopie[3] verallgemeinert auf beliebige Molekülarten, die zwischen zwei unterscheidbaren Zuständen A und B reversibel geschaltet werden können. Das Schalten der Farbstoffmoleküle in mindestens einen der beiden Zustände (z. B. vom Grundzustand A in den dunklen Zustand B) lässt sich durch Licht herbeiführen.
Die zu untersuchenden Präparate werden mit speziellen Molekülen, meist Fluoreszenzfarbstoffen, markiert. Die RESOLFT-Mikroskopie nutzt optisch getriebene, unterscheidbare Zustände in den Markermolekülen. Die Moleküle werden dabei zwischen mindestens zwei Zuständen hin- und hergeschaltet: Einem signalgebenden (hellen) Zustand A und einem dunklen Zustand B. Das Schalten der (Farbstoff-)Moleküle in mindestens einen der beiden Zustände (z. B. in den Zustand B) lässt sich durch Licht herbeiführen.
Das Präparat wird dabei inhomogen beleuchtet. Die Beleuchtungsintensität ist an mindestens einer vordefinierten Stelle sehr gering, idealerweise Null (also vollkommen dunkel). Nur an diesen dunklen Stellen werden die Moleküle also nicht in den Zustand B gebracht und verbleiben in A. Dieser Bereich lässt sich dann sehr klein (wesentlich kleiner als die klassische Beugungsgrenze) machen (siehe unten). Beim Detektieren des Signals (meist Fluoreszenzlicht) ist nun bekannt, dass es nur aus diesem kleinen Bereich kommen kann. Durch Verschieben des „A-Bereichs“ über das Präparat und Zusammensetzen der Teilbilder (Scannen) kann man daher Bilder mit der höheren Auflösung des „A-Bereichs“ erhalten.
Der Übergang der Markermoleküle aus den anderen Bildbereichen zurück in den Zustand A kann beispielsweise spontan oder durch Licht anderer Wellenlänge erfolgen. Die Markermoleküle müssen sich dafür mehrfach zwischen den Zuständen A und B hin- und herschalten lassen, um immer an gezielten Stellen den Zustand A und in den Nachbarbereichen den Zustand B zu behalten. Die Moleküle müssen in dem kleinen ausgewählten Bereich nicht notwendigerweise in den signalgebenden Zustand geschaltet werden. Auch eine Negativbildgebung ist möglich, bei der man aus dem kleinen Bereich gerade kein Signal bekommt. In diesem Fall ist eine mathematische Nachbearbeitung der Bilder nötig, um ein Positivbild zu erhalten.
Verkleinerung unter die Beugungsgrenze

Links: Bei niedriger Beleuchtungsintensität ist der (grüne) Bereich, der unter der Schwelle liegt, relativ groß.
Rechts: Durch Erhöhen der Intensität (ohne das Beleuchtungsprofil zu ändern) wird der Bereich, der unter der Schwelle liegt, viel kleiner.
Dies kann erzielt werden, da trotz der Beugungsgrenze der Bereich, in dem die Beleuchtungsintensität so niedrig ist, dass die Moleküle noch im Zustand A bleiben, beliebig klein gemacht werden kann (siehe Abbildung):
Um das Prinzip zu verstehen, machen wir zwei Annahmen:
- Das Präparat wird so beleuchtet, dass die Intensität an einer Stelle Null ist (rote Linie in der Abbildung). So eine Beleuchtung lässt sich z. B. durch Interferenzeffekte realisieren.
- Bei niedrigen Intensitäten (niedriger als die durch die blaue Linie in der Abbildung markierte Intensität) sind die Markermoleküle im (hellen) Zustand A. Bei höherer Intensität sind die Moleküle im (dunklen) Zustand B. Dies ist allerdings eine Vereinfachung – die Übergänge sind normalerweise nicht so abrupt.
Wird das Präparat nun mit niedriger Intensität beleuchtet (linke Abb.), ist der Bereich, in dem die Moleküle im Zustand A sind (grün markiert in der Abb.) relativ groß. Bereits durch Erhöhen der Intensität (also ohne dass die Form des Beleuchtungsprofils geändert wird), wird der Bereich, in dem die Intensität niedrig ist, kleiner (rechte Abb.). Folglich wird auch der Bereich, in dem die Moleküle im Zustand A sind, kleiner (grün markiert in der Abb.). Somit kommt das Fluoreszenzsignal nur noch aus einem sehr kleinen Bereich und es werden schärfere Abbildungen erhalten.
Varianten
Zum Schalten der Markermoleküle werden verschiedene Prozesse verwendet, die im Folgenden beschrieben werden. Allen Verfahren gemeinsam ist, dass der Marker mindestens zwischen zwei Zuständen hin- und hergeschaltet wird: einem signalgebenden (hellen) Zustand A und einem dunklen Zustand B.
STED-Mikroskopie
(Siehe auch den Artikel über das STED-Mikroskop).
Bei der STED-Mikroskopie (engl.: Stimulated Emission Depletion Microscopy)[1][2] kann ein Fluoreszenzfarbstoff in A zwischen seinem elektronischen Grundzustand und dem angeregten Zustand hin- und herpendeln und dabei fluoreszieren. In B wird der Farbstoff durch Stimulierte Emission permanent in seinem Grundzustand gehalten. Es gibt also zwei Konfigurationen der Fluoreszenzfarbstoffe: in A können sie fluoreszieren, in B nicht und die Voraussetzungen für RESOLFT sind vorhanden.
GSD-Mikroskopie
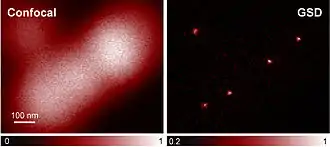
Auch bei der GSD-Mikroskopie (engl. Ground State Depletion Microscopy, dt. „Grundzustandsentvölkerungsmikroskopie“)[3] werden Fluoreszenzfarbstoffe als Marker verwendet. Der Farbstoff kann im hellen Zustand A zwischen dem Grundzustand und dem angeregten Zustand hin- und herpendeln und dabei fluoreszieren. Für den dunklen Zustand B wird der Grundzustand des Moleküls entvölkert: Das Molekül wird in einen langlebigen Zustand angeregt, von dem aus keine Fluoreszenz stattfindet. Solange sich das Molekül in dem langlebigen Dunkelzustand befindet, steht es nicht im Grundzustand zur Verfügung, kann also nicht angeregt werden und dementsprechend auch nicht fluoreszieren. Die Rückkehr in den hellen Zustand A erfolgt spontan. Oft handelt es sich bei dem langlebigen Zustand um einen sogenannten Triplettzustand. An Stickstoff-Fehlstellen-Zentren in Diamanten wurde eine Auflösung von bis zu 7,8 nm erreicht.[4] Im Vergleich mit einer herkömmlichen lichtmikroskopischen (konfokalen) Aufnahme wird der Auflösungsgewinn besonders deutlich (siehe Abb.).
SPEM und SSIM
SPEM (engl.: saturated pattern excitation microscopy)[5] und SSIM (engl.: saturated structured illumination microscopy)[6] sind RESOLFT-Verfahren, die zunächst Negativbilder aufnehmen und eine mathematische Bildrekonstruktion verwenden. Der Grundzustand tritt hier an die Stelle des dunklen Zustandes B und der erste angeregte Zustand ist der helle Zustand A.
RESOLFT-Mikroskopie mit schaltbaren Proteinen
Einige fluoreszierende Proteine können durch Licht geeigneter Wellenlänge ein- und ausgeschaltet und so für die RESOLFT-Mikroskopie verwendet werden.[7] Durch Bestrahlung mit Licht ändern sie ihre Struktur. Nur in einer dieser Strukturen ist das Protein zur Fluoreszenz fähig. Durch diese lichtinduzierte Strukturänderung können diese Proteine also von einem Zustand A in einen Zustand B geschaltet werden, von denen nur einer fluoresziert. Der Übergang vom Zustand B zurück in den Zustand A erfolgt entweder spontan oder auch durch Licht. Für das Schalten der Proteine werden im Vergleich zu den Verfahren STED und GSD nur sehr niedrige Lichtintensitäten benötigt (einige Watt pro Quadratzentimeter). In Kombination mit 4Pi Mikroskopie (4Pi-Mikroskop) wurden 2016 erstmals Aufnahmen mit isotroper Auflösung (< 40 nm) in lebenden Zellen und niedrigen Lichtintensitäten erstellt.[8]
RESOLFT-Mikroskopie mit schaltbaren organischen Farbstoffen
Ebenso wie in einigen Proteinen, können auch in bestimmten organischen Farbstoffen Strukturänderungen durch Licht induziert werden.[9] Die Fluoreszenzfähigkeit solche Farbstoffe lässt sich ebenso wie bei den Proteinen durch Licht an- und ausschalten. Auch hier werden nur relativ niedrige Intensitäten benötigt (einige hundert Watt pro Quadratzentimeter).
Verallgemeinerungen
Die beiden Zustände A und B müssen unterscheidbar sein, es muss aber nicht notwendigerweise Fluoreszenz involviert sein.[10] Das Schalten zwischen einem absorbierenden und einem nicht-absorbierenden Zustand oder einem streuenden und einem nicht-streuenden Zustand wäre ebenso möglich.
Quellen
- Stefan W. Hell: Microscopy and its focal switch. In: Nature Methods. Vol. 6, Nr. 1, 2009, S. 24–32, doi:10.1038/nmeth.1291.
- Stefan W. Hell: Far-Field Optical Nanoscopy. In: Science. Vol. 316, 2007, S. 1153–1158, doi:10.1126/science.1137395.
Weblinks
Einzelnachweise
- Stefan W. Hell, Jan Wichmann: Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. In: Optics Letters. Band 19, Nr. 11, 1994, S. 780–782, doi:10.1364/OL.19.000780.
- Thomas A. Klar, Stefan W. Hell: Subdiffraction resolution in far-field fluorescence microscopy. In: Optics Letters. Vol. 24, Nr. 14, 1999, S. 954–956, doi:10.1364/OL.24.000954.
- Die Methode wurde 1994 entwickelt, 1995 theoretisch beschrieben und 1997 experimentell demonstriert:
- Volker Dose: Peer review. In: EPL, A Letters Journal Exploring the Frontiers of Physics. Vol. 89, 2009, doi:10.1209/0295-5075/86/10000.
- Stefan W. Hell, M. Kroug: Ground-state-depletion fluorescence microscopy: a concept for breaking the diffraction resolution limit. In: Applied Physics B: Lasers and Optics. Vol. 60, Nr. 5, 1995, S. 495–497, doi:10.1007/BF01081333.
- Stefan Bretschneider, Christian Eggeling, Stefan W. Hell: Breaking the diffraction barrier in fluorescence microscopy by optical shelving. In: Physical Review Letters. Vol. 98, Nr. 5, 2007, S. 218103, doi:10.1103/PhysRevLett.98.218103.
- Eva Rittweger, Dominik Wildanger, Stefan W. Hell: Far-field fluorescence nanoscopy of diamond color centers by ground state depletion. In: EPL, A Letters Journal Exploring the Frontiers of Physics. Band 86, 2009, S. 14001, doi:10.1209/0295-5075/86/14001.
- Rainer Heintzmann, Thomas M. Jovin, Christoph Cremer: Saturated patterned excitation microscopy - a concept for optical resolution improvement. In: Journal of the Optical Society of America A. Vol. 19, Nr. 8, 2002, S. 1599–1609, doi:10.1364/JOSAA.19.001599.
- Mats G. L. Gustafsson: Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. In: Proceedings of the National Academy of Sciences. Vol. 102, Nr. 37, 2005, S. 13081–13086, doi:10.1073/pnas.0406877102.
- Michael Hofmann, Christian Eggeling, Stefan Jakobs, Stefan W. Hell: Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. In: Proceedings of the National Academy of Sciences. Vol. 102, Nr. 49, 2005, S. 17565–17569, doi:10.1073/pnas.0506010102.
- Ulrike Böhm, Stefan W. Hell, Roman Schmidt: 4Pi-RESOLFT nanoscopy. In: Nature Communications. Vol. 7, Nr. 10504, 2016, S. 1–8, doi:10.1038/ncomms10504.
- Mariano Bossi, Jonas Fölling, Marcus Dyba, Volker Westphal, Stefan W. Hell: Breaking the diffraction resolution barrier in far-field microscopy by molecular optical bistability. In: New Journal of Physics. Vol. 8, 2006, S. 275, doi:10.1088/1367-2630/8/11/275.
- Stefan W. Hell: Strategy for far-field optical imaging and writing without diffraction limit. In: Physics Letters A. Vol. 326, Nr. 1–2, 2004, ISSN 0375-9601, S. 140–145, doi:10.1016/j.physleta.2004.03.082.