Photochlorierung
Die Photochlorierung ist eine durch Licht ausgelöste chemische Reaktion, bei der in einer Kohlenwasserstoff-Verbindung Wasserstoff durch Chlor ersetzt wird, wobei als Koppelprodukt Chlorwasserstoff entsteht. Alternativ erfolgt eine radikalische Addition von Chlor an aromatische oder olefinische Kohlenwasserstoffe.

Die Photochlorierung wird neben der thermischen und der katalytischen Chlorierung auf industrieller Basis durchgeführt, meist in der Flüssigphase, zum Teil in Gegenwart inerter Lösungsmittel. Der Prozess ist exotherm und verläuft als Kettenreaktion, die durch die homolytische Spaltung von molekularem Chlor in Chlorradikale durch Ultraviolettstrahlung gestartet wird.
Die bei der Photochlorierung entstehenden chlorierten Kohlenwasserstoffe sind oft nur industrielle Zwischenprodukte und reagieren mit einer Vielzahl von Grundchemikalien zu Folgeprodukten wie Alkoholen, Mercaptanen, Aminen und Carbonsäuren. Die chemische Industrie nutzt niedermolekulare chlorierte Verbindungen wie Tetrachlorkohlenstoff als Lösungsmittel. Höhermolekulare Chloralkane dienen als Insektizide, als Flammschutzmittel oder als Weichmacher in Kunststoffen und Beschichtungen. Chlorierte Kohlenwasserstoffe dienen weiterhin als Zwischenprodukt in der chemischen Industrie zur Herstellung von Silikonen oder Waschmitteln.
Geschichte

Bei der Chlorierung handelt es sich um eine der ältesten bekannten Substitutionsreaktionen der Chemie. Der französische Chemiker Jean-Baptiste Dumas untersuchte bereits um 1830 die Substitution von Wasserstoff durch Chlor bei der Einwirkung von Chlor auf Kerzenwachs und Essigsäure.[1] Er wies dabei nach, dass sich für jedes in einen Kohlenwasserstoff eingebrachte Mol Chlor ein Mol Chlorwasserstoff bildete, und bemerkte die Lichtempfindlichkeit dieser Reaktion.[2]
Die ersten Arbeiten zum Einfluss des Lichts auf die Geschwindigkeit chemischer Reaktionen stammen von Theodor Grotthuß. Grotthuß veröffentlichte bereits 1819 eine Abhandlung über die chemische Wirksamkeit des Lichts und formulierte das photochemische Absorptionsgesetz. Demzufolge ruft in einem physikalisch-chemischen System nur derjenige Bruchteil der einfallenden Strahlung eine Wirkung hervor, der von diesem System absorbiert wird; reflektierte und transmittierte Strahlung bleibt ohne Wirkung.[3]
Durch die im Jahr 1900 veröffentlichte Arbeit von Max Planck war bekannt, dass Licht aus diskreten Quanten besteht.[4] Die Anregung einer einzelnen chemischen Reaktion durch ein Lichtquant konnte dadurch erklärt werden, jedoch nicht die Quantenausbeute von Reaktionen wie der Photochlorierung. Die Idee, dass es sich bei diesen Reaktionen um Kettenreaktionen handeln könnte, stammt von Max Bodenstein aus dem Jahr 1913. Er nahm an, dass bei der Reaktion zweier Moleküle nicht nur das Endprodukt der Reaktion entstehen kann, sondern auch instabile, reaktive Zwischenstufen, die eine Kette fortführen können.[5]
Wegen der Bedeutung der Reaktion für das Verständnis der Chemie, der Substitutionsmuster und der entstehenden Derivate untersuchten Chemiker die Reaktion eingehend. In die chemisch-industrielle Praxis konnte die Photochlorierung jedoch erst überführt werden, als gegen Ende des neunzehnten Jahrhunderts billiges Chlor aus der Chloralkali-Elektrolyse zur Verfügung stand.[6]
Eine erste Anwendung fanden chlorierte Alkane in Rachensprays. Diese enthielten um 1914 bis 1918 in relativ großen Mengen chlorierte Alkane als Lösungsmittel für Chloramin T. Die Sharpless Solvents Corporation nahm 1929 die erste industrielle Photochlorierungsanlage zur Chlorierung von Pentan in Betrieb.[7] Die kommerzielle Produktion von Chlorparaffinen zur Verwendung als Hochdruckadditive in Schmierstoffen begann um 1930.[8] Um das Jahr 1935 lief das Verfahren technisch stabil und kommerziell erfolgreich.[7]
Aber erst in den Jahren nach dem Zweiten Weltkrieg begann ein größerer Aufbau von Photochlorierungskapazität. Im Jahr 1950 produzierten die Vereinigten Staaten bereits über 800.000 Tonnen chlorierter Paraffinkohlenwasserstoffe. Die Hauptprodukte waren Ethylchlorid, Tetrachlorkohlenstoff und Methylenchlorid.[9] Wegen der Bedenken hinsichtlich der gesundheitlichen und umweltrelevanten Probleme wie dem Ozonabbauverhalten leichtflüchtiger Chlorverbindungen entwickelte die chemische Industrie alternative Verfahren, die ohne chlorierte Verbindungen auskamen. Durch den dadurch bedingten Ersatz der chlorierten durch nicht-chlorierte Produkte sank die weltweite Produktionsmenge im Laufe der Jahre beträchtlich.[8][10]
Hintergrund

Da nur absorbiertes Licht eine photochemische Primärreaktion auslöst, muss bei jeder photochemischen Reaktion einer der Reaktionspartner dieses Licht absorbieren. Im Falle der Photochlorierung ist das Chlor der absorbierende Reaktionspartner. Chlor absorbiert Licht in einem Wellenlängenbereich von etwa 250 bis 450 Nanometern, entsprechend einer Absorption im langwelligen ultravioletten und im sichtbaren violetten Spektralbereich.[11] Für die homolytische Spaltung von Chlor ist eine Energie von 244 Kilojoule pro Mol notwendig.[12]
Gemäß dem photochemischen Äquivalenzgesetz verursacht jedes absorbierte Photon eine photochemische Primärreaktion.[13]

mit NA als Avogadro-Konstante (NA = 6,022 1023 mol−1), dem planckschen Wirkungsquantum (h = 6,626 10−34 Js) und der Lichtfrequenz mit der Einheit s−1.



Über die Beziehung:

mit für die Lichtgeschwindigkeit (c = 299.792.458 ms−1) ergibt sich


und damit für die Wellenlänge in der Einheit nm:


Durch Einsetzen der Werte ergibt sich ein Wert für die Wellenlänge von 491 Nanometern, die das eingestrahlte Licht maximal aufweisen darf, um eine Spaltung von Chlor zu bewirken. Das Absorptionsmaximum von Chlor liegt bei etwa 340 Nanometern.[14] Licht dieser Wellenlänge strahlt etwa eine Energie von 377 Kilojoule pro Mol ein und ist damit mehr als ausreichend für eine Photolyse des Chlors.
Für photochemische Prozesse ist es wichtig, wie sich die Zahl der umgesetzten Moleküle zur Zahl der absorbierten Lichtquanten verhält. Dieses Verhältnis wird als Quantenausbeute (QA) bei einer bestimmten Lichtwellenlänge bezeichnet. Das Verhältnis von umgesetzten Molekülen des lichtabsorbierenden Stoffs zur Anzahl der absorbierten Photonen berechnet sich als:



Die Quantenausbeute sollte beim photochemischen Primärprozess, der Absorption des Lichtquants, den Wert 1 nicht übersteigen. Bei der Photochlorierung ist dieses Verhältnis jedoch nicht gleich oder kleiner 1, sondern wegen der Sekundärprozesse, bei denen die gleichen Stoffe entstehen wie beim photochemischen Primärprozess, oft beträchtlich größer.[14]
Reaktion
Substitutionsreaktion
Die Substitution der Wasserstoffatome in einem Kohlenwasserstoff erfolgt rein statistisch, wobei tertiäre Wasserstoffatome schneller reagieren als sekundäre und diese schneller als primäre. Bei einer Temperatur von 30 °C verhalten sich die relativen Reaktionsgeschwindigkeiten von primären, sekundären und tertiären Wasserstoffatomen etwa wie 1 zu 3,25 zu 4,43.[14] Eine Umlagerung des Kohlenstoffgerüsts findet nicht statt, aber es werden immer alle möglichen Monochloride gebildet.[15]
Die Reaktion läuft bei Belichtung unter Beteiligung von Alkyl- und Chlorradikalen als Kettenträger nach dem folgenden Schema ab:





Der Kettenabbruch erfolgt durch Rekombination von Chlorradikalen zu molekularem Chlor an der Gefäßwand.[16] Verunreinigungen wie Sauerstoff, der in elektrochemisch gewonnenem Chlor vorkommt, verursachen ebenfalls einen Kettenabbruch.
Bei der Photochlorierung erfolgt keine Umlagerung der Kohlenstoffkette und es entstehen alle möglichen Monochloride sowie mehrfach chlorierte Verbindungen.[15] Zielprodukte der Photochlorierung sind jedoch meist die monosubstituierten Chlorkohlenwasserstoffe. Die Bildung mehrfach substituierter Produkte lässt sich in Grenzen durch das Arbeiten mit einem hohen Überschuss an Kohlenwasserstoffen oder durch Verdünnung des Chlors mit Stickstoff vermindern.
Die Selektivität der Photochlorierung hinsichtlich einer Substitution von primären, sekundären oder tertiären Wasserstoffatomen kann durch die Interaktion des Chlorradikals mit dem Lösungsmittel, etwa Benzol, tert-Butylbenzol oder Kohlenstoffdisulfid, gesteuert werden.[17] Durch die Bildung eines Komplexes aus Benzol und dem Chlorradikal vermindert sich die Reaktivität gegenüber einem freien Chlorradikal, die Selektivität der Photochlorierung erhöht sich dadurch.[18] Die durch die Wahl des Lösungsmittels erhaltene Bandbreite des Verhältnisses von substituierten primären zu sekundären Wasserstoffatomen liegt bei 1 : 3 bis 1 : 31.[19] Bei höheren Temperaturen gleichen sich die Reaktionsgeschwindigkeiten von primären, sekundären und tertiären Wasserstoffatomen an. Daher wird die Photochlorierung meist bei tieferen Temperaturen durchgeführt.[15]



Reaktionstechnik

Ausgangsstoffe (Reaktanten) der Photochlorierung können sowohl gasförmige als auch flüssige Kohlenwasserstoffe sein. Bei flüssigen Ausgangsstoffen wird Chlor unter Rühren eingeleitet. Reine Gasreaktionen, etwa die Photochlorierung von Methan, sind prinzipiell möglich; der Temperaturanstieg in einer Gasphasenreaktion muss weitgehend durch die spezifischen Wärmekapazitäten der beteiligten Gase aufgenommen werden, was den Umsatz limitiert.[21] Generell ist es notwendig, die Reaktanten nahe an die Lichtquelle zu bringen, um eine möglichst hohe Lichtausbeute zu erhalten. Dazu kann das Reaktionsgemisch direkt oder in einem durchströmten Seitenarm eines Reaktors mit einer geeigneten Lichtquelle bestrahlt werden. Gasförmige Kohlenwasserstoffe werden in ein inertes Lösungsmittel eingeleitet und dort unter Bestrahlung mit Chlor zur Reaktion gebracht.[22]
Ein Nachteil photochemischer Prozesse ist der geringe Wirkungsgrad der Umwandlung elektrischer Energie in Strahlungsenergie der benötigten Wellenlänge. Neben der Strahlung erzeugen Lichtquellen viel Wärme, die wiederum Kühlleistung benötigt. Außerdem strahlen die meisten Lichtquellen polychromatisches Licht aus, obwohl nur monochromatisches Licht benötigt wird.[23] Eine hohe Quantenausbeute gleicht diese Nachteile jedoch aus. Die Quantenausbeute für die Photochlorierung des n-Heptans beträgt zum Beispiel etwa 7000.[24] In technischen Anlagen zur Photochlorierung beträgt die Quantenausbeute etwa 100. Im Gegensatz zur thermischen Chlorierung, welche die entstandene Reaktionswärme nutzen kann, muss bei der photochemischen Arbeitsweise die Energie zur Aufrechterhaltung der Reaktion ständig nachgeliefert werden.[22]

Die Gegenwart von Inhibitoren wie beispielsweise Sauerstoff oder von Stickoxiden muss vermieden werden. Zu hohe Chlorkonzentrationen führen zu einer zu starken Absorption in der Nähe der Lichtquelle und wirken sich nachteilig aus.[19] Ein Arbeiten bei niedrigen Temperaturen ist vorteilhaft, da Nebenreaktionen vermieden werden, da die Selektivität erhöht wird und da gasförmige Reaktionspartner weniger aus einem Lösungsmittel ausgetrieben werden, was die Ausbeute erhöht. Die Ausgangsstoffe können vor der Reaktion zum Teil so weit abgekühlt werden, dass die Reaktionswärme ohne weitere Kühlung des Gemischs aufgenommen wird. Bei gasförmigen oder leichtsiedenden Ausgangsstoffen ist ein Arbeiten unter Druck erforderlich. Auf Grund der Vielzahl von möglichen Rohstoffen ist eine große Anzahl von Prozessen beschrieben worden.[25][26] Die Photochlorierung wird meist in einem Rührkesselreaktor, einem Blasensäulenreaktor oder einem Rohrreaktor durchgeführt, denen je nach Zielprodukt weitere Aufarbeitungsstufen folgen.[27] Im Falle des Rührkesselreaktors wird die Lampe, die in der Regel als länglicher Zylinder geformt ist, mit einem Kühlmantel versehen und in die Reaktionslösung eingetaucht. Rohrreaktoren sind Quarz- oder Glasrohre, die von außen bestrahlt werden. Die Rührkesselvariante besitzt den Vorteil, dass kein Licht an die Umgebung verloren geht. Jedoch fällt die Lichtintensität schnell mit dem Abstand zur Lichtquelle aufgrund von Adsorption durch die Reaktanten ab.[22]
Der Einfluss der Strahlung auf die Reaktionsgeschwindigkeit lässt sich oft durch ein Potenzgesetz auf Basis der Quantenstromdichte, also der Mole Lichtquanten (früher in der Einheit Einstein gemessen) pro Fläche und Zeit, darstellen. Ein Ziel bei der Auslegung von Reaktoren ist es daher, die wirtschaftlich günstigste Dimensionierung hinsichtlich einer Optimierung der Quantenstromdichte zu bestimmen.[28]
Produkte
Chlorierte Produkte können über eine Vielzahl von Reaktionen in weitere Zwischen- und Endprodukte überführt werden, etwa durch Hydrolyse in Alkohole oder durch Reaktion mit Alkalicyaniden in Nitrile, die mit Wasser zu Carbonsäuren hydrolysiert oder mit Wasserstoff zu Aminen reduziert werden können. Durch Umsatz mit metallischem Magnesium in Grignard-Reaktionen lassen sich über die Zwischenstufe von Alkyl-Magnesiumhalogeniden Kohlenstoffgerüste aufbauen.[29] In Friedel-Crafts-Alkylierungen dienen Chloralkane zur Darstellung von Alkylaromaten.[30]
Chlorparaffine

Chlorparaffine lassen sich durch Photochlorierung aus Alkanen darstellen. Gegenüber einer thermischen Chlorierung ist die Gefahr der Bildung von Folgeprodukten durch Thermolyse, zum Beispiel durch Abspaltung von Chlorwasserstoff, nur sehr gering. Durch den radikalischen Reaktionsverlauf ist die Selektivität gering und es entstehen Gemische mehrerer Chlorparaffine mit komplexer Zusammensetzung. Der Chlorierungsgrad variiert, die genaue Zusammensetzung der entstehenden Produktgemische ist oft nicht bekannt.[8] Die Weltjahresproduktion betrug 1985 300.000 Tonnen; seither sind die Produktionsmengen in Europa und Nordamerika rückläufig.[31] In China dagegen stieg die Produktion stark an. China produzierte 2007 über 600.000 Tonnen Chlorparaffine, im Jahr 2004 lag die Menge noch unter 100.000 Tonnen.[32]
Die Chlorparaffine besitzen die allgemeine Summenformel CxH(2x−y+2)Cly und werden in drei Gruppen eingeteilt. Die niedermolekularen Chlorparaffine sind die kurzkettigen Chlorparaffine (Short Chain Chloroparaffins (SCCP)) mit 10 bis 13 Kohlenstoffatomen, es folgen die mittelkettigen Chlorparaffine (Medium Chain Chloroparaffins (MCCP)) mit Kohlenstoffkettenlängen von 14 bis 17 Kohlenstoffatomen und die langkettigen Chlorparaffine (Long Chain Chloroparaffins (LCCP)), wobei die Kohlenstoffkettenlänge größer als 17 Kohlenstoffatome ist. Bei etwa 70 % der hergestellten Chlorparaffine handelt es sich um MCCP mit einem Chlorierungsgrad von 45 bis 52 %. Die restlichen 30 % verteilen sich zu gleichen Teilen auf SCCP und LCCP.[8]
Die Short Chain Chloroparaffins weisen eine hohe Toxizität auf und reichern sich leicht in der Umwelt an. Die Europäische Union hat die SCCP als Kanzerogene der Kategorie III eingestuft und ihre Verwendung eingeschränkt.[33]
Benzylchlorid, Benzalchlorid und Benzotrichlorid
Durch die Photochlorierung der Seitenkette des Toluols lassen sich die mono- bis trichlorierten Produkte herstellen, deren wichtigster Vertreter das Benzylchlorid ist. Überführt in den Benzylalkohol dient es als Zwischenprodukt für die Herstellung von Weichmachern. Durch die Überführung in das Benzylcyanid unter nachfolgender Hydrolyse wird schließlich Phenylessigsäure gewonnen.[34][35]
Das disubstituierte Benzalchlorid ist der Rohstoff für die Gewinnung von Benzaldehyd. Als Reinstoff wird Benzaldehyd eingesetzt, um Lebensmitteln einen Mandelgeruch zu verleihen.[36] Als Zwischenprodukt dient es der Herstellung von Malachitgrün und anderen Farbstoffen.[37] Das trisubstituierte Benzotrichlorid dient durch Hydrolyse der Synthese von Benzoylchlorid:[38]


Durch Reaktion mit Alkoholen lässt sich Benzoylchlorid in die entsprechenden Ester überführen. Mit Natriumperoxid setzt es sich zu Benzoylperoxid um, einem Radikalstarter für Polymerisationen. Die Atomökonomie dieser Synthesen ist jedoch gering, da dabei stöchiometrische Mengen Salze anfallen.
Chlormethane
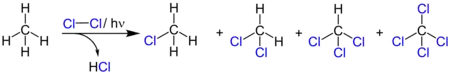
Ein Beispiel für eine Photochlorierung bei tiefen Temperaturen und unter Normaldruck ist die Chlorierung von Methylchlorid zu Methylenchlorid. Das verflüssigte Methylchlorid, welches bei −24 °C siedet, wird dazu im Dunkeln mit Chlor gemischt und anschließend mit einer Quecksilberdampflampe bestrahlt. Das entstehende Methylenchlorid hat einen Siedepunkt von 41 °C und wird später destillativ vom Methylchlorid getrennt.[39]
Die Photochlorierung von Methan weist eine niedrigere Quantenausbeute auf als die Chlorierung von Methylenchlorid. Durch den dadurch notwendigen hohen Lichteinsatz erfolgt eine direkte Weiterchlorierung der Zwischenprodukte, so dass dabei hauptsächlich Tetrachlorkohlenstoff entsteht.[39]
Monochlornonan und -dodecan

Monochlornonan reagiert in einer Friedel-Crafts-Alkylierung mit Phenol zu Nonylphenol und kann durch Ethoxylierung weiter zu Nonylphenolethoxylaten umgesetzt werden. Diese nichtionische Tenside werden als Emulgatoren und als Wasch- und Reinigungsmittel eingesetzt. Aufgrund der xenoestrogenen Eigenschaften wurde die Verwendung von Nonylphenolethoxylaten und Nonylphenolen in der EU stark eingeschränkt.[40]
Ein weiteres Zielprodukt ist das Monochlordodecan, das mit Benzol ebenfalls in einer Friedel-Crafts-Alkylierung zu einem Waschmittelrohstoff, dem linearen Alkylbenzol, reagiert, welches zu Natriumdodecylbenzolsulfonat weiterverarbeitet wird. Bei der Photochlorierung des Dodecans lässt sich durch die Wahl eines geeigneten Lösungsmittels wie etwa Benzol die Bildung unerwünschter 1-Isomere unterdrücken.[18] Mittlerweile erfolgt die Produktion des Alkylbenzols meist über die Fluorwasserstoff-katalysierte Reaktion von 1-Dodecen mit Benzol.[41]
Chlorierte Polymere
Polyethylen, in Tetrachlorkohlenstoff gelöst, lässt sich photochemisch in einer polymeranalogen Reaktion in ein chloriertes Polyolefin überführen, welches als Schlagzähmodifier für die Verbesserung der Kerbschlagzähigkeit von Polyvinylchlorid verwendet wird. Durch Photochlorierung lassen sich Polyvinylchlorid-Folien bei Raumtemperatur weiter chlorieren. Dabei werden die nicht-chlorierten Methylengruppen des Polymers chloriert. Die Quantenausbeute liegt bei Reaktionen zwischen Polymeren in der Festphase und Chlor typischerweise in der Gegend von 1, da eine Kettenreaktion in diesem Fall nicht oder nur eingeschränkt möglich ist.[42]
Polyolefin-Membranen aus Polyethylen-, Polypropylen- und Polystyrol-Folien lassen sich in der festen Phase durch Photochlorierung zu Membranen mit einem Chlorgehalt von bis zu 12 % chlorieren. Dadurch lassen sich die Gaspermeation, die Benetzbarkeit und die Wasserdurchlässigkeit verbessern.[43]
Hexachlorcyclohexan

Bei der Photochlorierung von Benzol entstehen von den acht möglichen Isomeren die vier α-, β-, γ-, δ-Hexachlorcyclohexan-Isomere in größerem Maßstab. Diese liegen alle in einer Sesselkonformation vor und unterscheiden sich durch die Besetzung axialer und äquatorialer Positionen im Molekül. Eine insektizide Wirkung zeigt nur das γ-Isomer, das drei Chloratome in axialer und drei in äquatorialer Position enthält und in Konzentrationen von 10 bis 18 % entsteht.[20] Es wird durch Extraktionsverfahren von den anderen Isomeren abgetrennt, die durch Dehydrochlorierung zu Trichlorbenzol weiterverarbeitet werden. Das technisch reine γ-Isomer wird unter Handelsnamen wie Lindan und Gammexan außerhalb der EU noch als Insektizid verwendet.[20]
Sonstige Produkte
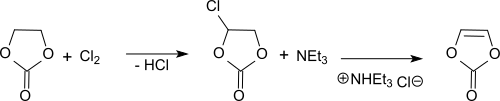
Durch Photochlorierung von Ethylencarbonat lässt sich Vinylencarbonat über die Stufe des Monochlorethylencarbonat unter anschließender Dehydrochlorierung, etwa mit Triethylamin (NEt3), gewinnen. Vinylencarbonat ist ein reaktives Monomer für die Homopolymerisation und Copolymerisation, etwa mit Isobutylvinylether.[44]
Geringe Spuren von Trichlorsilan in ultrareinem Tetrachlorsilan können durch Photochlorierung entfernt werden.[27]
Verwertung von Chlorwasserstoff
Chlorwasserstoff kann zur weiteren Chlorierung, etwa durch Additionsreaktion von Chlorwasserstoff an eine olefinische Doppelbindung, durch Veresterung von Alkoholen oder durch Oxychlorierung von Alkanen und Olefinen, verwendet werden. Der bei der Photochlorierung von Methan entstehende Chlorwasserstoff kann etwa durch Veresterung mit Methanol selektiv zu Methylchlorid umgesetzt werden.[45] Bei erhöhter Temperatur entsteht unter Katalyse auch Methylenchlorid.
Verfahrensvarianten
Strahlenchemische Chlorierung

Statt ultravioletten Licht wird auch Gammastrahlung zum Radikalkettenstart bei der Chlorierung verwendet. Die trockene Nachchlorierung von PVC im Wirbelbett, die so genannte PC-Chlorierung, führte das Chemiekombinat Bitterfeld strahlenchemisch durch.[46] Das langlebige Isotop Cobalt-60 wurde dafür als Gammastrahlungsquelle verwendet.[47]
Sulfochlorierung
Unter fast identischen Bedingungen und gleicher Reaktionsführung wie die herkömmliche Photochlorierung läuft die 1936 von Cortes F. Reed zuerst beschriebene Sulfochlorierung ab.[48] Neben Chlor wird dabei noch Schwefeldioxid in das Reaktionsgemisch eingeführt. Als Produkte entstehen Alkylsulfonylchloride, die zu Tensiden weiterverarbeitet werden.[49]
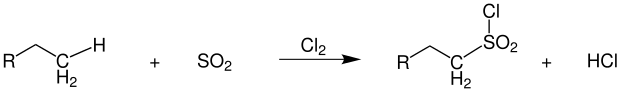 Übersichtsreaktion der Reed-Reaktion
Übersichtsreaktion der Reed-Reaktion
Als Koppelprodukt entsteht wie bei der Photochlorierung Chlorwasserstoff. Da eine direkte Sulfonierung der Alkane kaum möglich ist, hat sich diese Reaktion als nützlich erwiesen. Durch das direkt am Schwefel gebundene Chlor sind die entstehenden Produkte äußerst reaktiv. Als Nebenprodukte finden sich im Reaktionsgemisch Alkylchloride, die durch reine Photochlorierung entstehen, sowie mehrfach sulfochlorierte Produkte.[50]
Photobromierung
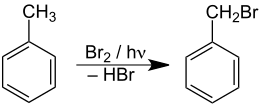
Die Photobromierung mit elementarem Brom verläuft ebenfalls nach einem radikalischen Mechanismus analog zur Photochlorierung. Bei der Anwesenheit von Sauerstoff erfolgt teilweise eine Oxidation des entstehenden Bromwasserstoffs zum Brom, was zu einer erhöhten Ausbeute führt.[51] Wegen der leichteren Dosierbarkeit des elementaren Broms und der höheren Selektivität der Reaktion wird für Arbeiten im Labormaßstab die Photobromierung der Photochlorierung vorgezogen. Für industrielle Anwendungen ist Brom, das nur in geringen Mengen im Meerwasser enthalten ist und aus diesem durch Oxidation mit Chlor in Freiheit gesetzt wird, jedoch meist zu teuer.[52][53] Anstelle von elementarem Brom eignet sich auch N-Bromsuccinimid als Bromierungsmittel.[54] Die Quantenausbeute der Photobromierung ist meist wesentlich geringer als die der Photochlorierung.
Literatur
- Dieter Wöhrle, Michael Tausch, Wolf-Dieter Stohrer: Photochemie: Konzepte, Methoden, Experimente. Wiley & Sons, 1998, ISBN 3-527-29545-3.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 3-13-201904-6.
- Mario Schiavello (Hrsg.): Photoelectrochemistry, Photocatalysis and Photoreactors Fundamentals and Developments. Springer Netherlands, 2009, ISBN 978-90-481-8414-9.
Weblinks
Einzelnachweise
- Jean-Baptiste Dumas: Ueber die Einwirkung des Chlors auf den aus essigsauren Salzen entstehenden Kohlenwasserstoff. In: Annalen der Chemie und Pharmacie. 33, 1840, S. 187–189, doi:10.1002/jlac.18400330205.
- Jean-Baptiste Dumas: Über das Gesetz der Substitution und die Theorie der Typen. In: Lieb. Ann. Vol. 33, 1840, S. 259–300.
- Theodor Von Grotthuss: Auszug aus vier Abhandlungen Physikalisch-chemischen Inhalts. In: Annalen der Physik und der physikalischen Chemie. 61, 1819, S. 50–74, doi:10.1002/andp.18190610105.
- Max Planck: Zur Theorie des Gesetzes der Energieverteilung im Normalspectrum. In: Verhandlungen der Deutschen physikalischen Gesellschaft. 2, Nr. 17, 1900, S. 245, Berlin (vorgetragen am 14. Dezember 1900); (PDF) (Memento vom 7. August 2015 im Internet Archive)
- Max Bodenstein: Photochemische Kinetik des Chlorknallgases. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 19, 1913, S. 836–856, doi:10.1002/bbpc.19130192104.
- Franz Rudolf Minz, Reinhard Schliebs: Moderne Verfahren der Großchemie: Chlor und Natronlauge. In: Chemie in unserer Zeit. 12. Jahrg. 1978, Nr. 5, ISSN 0009-2851, S. 135–145.
- Wilhelm Hirschkind: Chlorination of Saturated Hydrocarbons. In: Industrial & Engineering Chemistry. 41, 1949, S. 2749–2752, doi:10.1021/ie50480a021.
- United Nations Environment Programme, International Labour Organisation, World Health Organisation, International Programme on Chemical Safety, Environmental Health Criteria 181: CHLORINATED PARAFFINS.
- Earl T. McBee, Ogden R Pierce: Halogenation. In: Industrial & Engineering Chemistry. 46, 1954, S. 1835–1841, doi:10.1021/ie50537a031.
- Martin Dameris, Thomas Peter, Ulrich Schmidt, Reinhard Zellner: Das Ozonloch und seine Ursachen. In: Chemie in unserer Zeit. 41, 2007, S. 152–168; doi:10.1002/ciuz.200700418.
- Hans Von Halban: Die Lichtabsorption des Chlors. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 28, 1922, S. 496–499, doi:10.1002/bbpc.19220282304.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 71.
- Arthur John Allmand: Part I.—Einstein’s law of photochemical equivalence. Introductory address to Part I. In: Trans. Faraday Soc. 21, 1926, S. 438, doi:10.1039/TF9262100438.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 3-13-201904-6, S. 91.
- Keith U. Ingold, J. Lusztyk, K. D. Raner: The unusual and the unexpected in an old reaction. The photochlorination of alkanes with molecular chlorine in solution. In: Accounts of Chemical Research. 23, 1990, S. 219, doi:10.1021/ar00175a003.
- Max Bodenstein: Sitzung vom 15. Dezember 1930. Berichte der deutschen chemischen Gesellschaft (A and B Series) 64.1 (1931): A1–A4.
- Glen A. Russell: Solvent Effects in the Reactions of Free Radicals and Atoms. III. Effects of Solvents in the Competitive Photochlorination of Hydrocarbons and Their Derivatives. In: Journal of the American Chemical Society. 80, 1958, S. 4997–5001, doi:10.1021/ja01551a057.
- D. J. Hurley, R. W. Rosenthal, R. C. Williamson: Effect of Chlorination Conditions on Preparation and Isomer Distribution of Linear Detergent Alkylate. In: Industrial & Engineering Chemistry Product Research and Development. 4, 1965, S. 22, doi:10.1021/i360013a007.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 3-13-201904-6, S. 95.
- Richard Wegler (Hrsg.): Chemie der Pflanzenschutz und Schädlingsbekämpfungsmittel. Band 1, Springer-Verlag, 1970, ISBN 3-642-46212-X, S. 129–132.
- Mario Schiavello (Hrsg.): Photoelectrochemistry, Photocatalysis and Photoreactors Fundamentals and Developments. Springer Netherlands, 2009, ISBN 978-90-481-8414-9, S. 564.
- Martin Fischer: Industrial Applications of Photochemical Syntheses. In: Angewandte Chemie International Edition in English. 17, 1978, S. 16–26, doi:10.1002/anie.197800161.
- Dieter Wöhrle, Michael W. Tausch, Wolf-Dieter Stohrer: Photochemie: Konzepte, Methoden, Experimente. Wiley & Sons, 1998, ISBN 3-527-29545-3, S. 271–275.
- Joachim Stauff, H. J. Schumacher: Apparatur zur Untersuchung von Lichtreaktionen der Halogene mit organischen Substanzen: Die Lichtreaktion zwischen Chlor und n‐Heptan. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 48, 1942, S. 271–278, doi:10.1002/bbpc.194200006.
- Patent US1379367: Process of Chlorination. Veröffentlicht am 24. Mai 1921, Erfinder: F. Sparre, W. E. Masland.
- Patent US1459777: Process and Apparatus for the Chlorination of Methane. Veröffentlicht am 14. Februar 1920, Erfinder: R. Leiser, F. Ziffer.
- David A. Mixon, Michael P. Bohrer, Patricia A. O’Hara: Ultrapurification of SiCl4 by photochlorination in a bubble column reactor. In: AIChE Journal. 36, 1990, S. 216–226, doi:10.1002/aic.690360207.
- H. Hartig: Einfache Dimensionierung, photochemischer Reaktoren. In: Chemie Ingenieur Technik – CIT. 42, 1970, S. 1241–1245, doi:10.1002/cite.330422002.
- Victor Grignard: Sur quelques nouvelles combinaisons organométalliques du magnèsium et leur application à des synthèses d’alcools et d’hydrocarbures. In: CR Hebd. Séances Acad. Sci. Ser. C, 130, 1900, S. 1322–1324, Digitalisat auf Gallica, dt. Über einige neue metallorganische Verbindungen von Magnesium und deren Anwendung auf Synthesen von Alkoholen und Kohlenwasserstoffen
- Charles Friedel, James Mason Crafts: Sur une nouvelle méthode générale de synthèse d’hydrocarbures, d’acétones, etc. In: Compt. Rend. 84, S. 1392 & 1450.
- Heinz Strack: Chlorinated paraffins. In: Ullmann’s Encyclopedia of Industrial Chemistry. Vol. A6, Weinheim, 1986, VCH Verlagsgesellschaft, S. 323–330.
- Heidelore Fiedler (Hrsg.): Chlorinated Paraffins. In: The Handbook of Environmental Chemistry. Springer-Verlag, 2010, ISBN 978-3-642-10760-3, S. 8.
- Richtlinie 2002/45/EG des Europäischen Parlaments und des Rates vom 25. Juni 2002 zur 20. Änderung der Richtlinie 76/769/EWG des Rates hinsichtlich der Beschränkungen des Inverkehrbringens und der Verwendung gewisser gefährlicher Stoffe und Zubereitungen (kurzkettige Chlorparaffine), abgerufen am 5. März 2016
- Roger Adams, A. F. Thal: Benzyl Cyanide. In: Organic Syntheses. 2, 1922, S. 9, doi:10.15227/orgsyn.002.0009.
- Roger Adams, A. F. Thal: Phenylacetic Acid [α-Toluic acid]. In: Organic Syntheses. 2, 1922, S. 63, doi:10.15227/orgsyn.002.0063.
- Final Report on the Safety Assessment of Benzaldehyde. In: International Journal of Toxicology. 25, 2006, S. 11–27, doi:10.1080/10915810600716612.
- Siegfried Hauptmann: Organische Chemie. 2. Auflage. VEB Deutscher Verlag für Grundstoffindustrie, Leipzig, 1985, ISBN 3-342-00280-8, S. 757.
- Barbara Elvers (Hrsg.): Ullmann’s Encyclopedia of Industrial Chemistry: 7th Edition. Wiley-VCH, 2002, ISBN 3-527-30385-5, S. 139.
- Eugen Müller (Hrsg.), E. Forche, W. Hahn: Methoden der organischen Chemie Band V/3: Halogenverbindungen. Fluorverbindungen. Herstellung, Reaktivität und Umwandlung. Chlorverbindungen. Thieme Verlag, 1962, S. 571–573.
- Richtlinie 2003/53/EG des Europäischen Parlaments und des Rates vom 18. Juni 2003 zur 26. Änderung der Richtlinie 76/769/EWG des Rates über Beschränkungen des Inverkehrbringens und der Verwendung gewisser gefährlicher Stoffe und Zubereitungen (Nonylphenol, Nonylphenolethoxylat und Zement), abgerufen am 25. März 2016.
- Marion L. Sharrah, Geo. C. Feighner: Synthesis of Dodecylbenzen – Synthetic Detergent Intermediate. In: Industrial & Engineering Chemistry. 46, 1954, S. 248–254, doi:10.1021/ie50530a020.
- C. Decker, M. Balandier, J. Faure: Photochlorination of Poly(vinyl Chloride). I. Kinetics and Quantum Yield. In: Journal of Macromolecular Science: Part A – Chemistry. 16, 2006, S. 1463–1472, doi:10.1080/00222338108063248.
- Tsutomu Nakagawa, Sumio Yamada: Modification of polyolefin films by photochlorination. In: Journal of Applied Polymer Science. 16, 1972, S. 1997–2012, doi:10.1002/app.1972.070160813.
- Rolf C. Schulz, Rainer Wolf: Copolymerisation zwischen Vinylencarbonat und Isobutylvinyläther. In: Kolloid-Zeitschrift & Zeitschrift für Polymere. 220, 1967, S. 148–151, doi:10.1007/BF02085908.
- Ernst Bartholomé, Ernst Biekert, Heinrich Hellmann: Umwelt- und Arbeitsschutz. (Ullmanns Encyklopädie der technischen Chemie Band 6). Wiley-VCH, 4. Auflage, 1981, ISBN 3-527-20006-1, S. 206.
- R. Newe, P. Schmidt, K. Friese, B. Hösselbarth: Das Verfahren der strahlenchemischen Chlorierung von Polyvinylchlorid. In: Chemische Technik. 41(4), 1989, S. 141–144.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.), Otto Bayer, Hans Meerwein, Karl Ziegler: Methoden der organischen Chemie. V/3 Fluorine and Chlorine Compounds . Thieme Verlag, Stuttgart 1962, ISBN 3-13-203004-X, S. 524.
- Patent US2046090: Method of halogenating compounds and product resulting therefrom. Veröffentlicht am 30. Juni 1936, Erfinder: Cortes F. Reed.
- Patent US2174492: Preparation of alkane sulphonyl chlorides. Veröffentlicht am 26. September 1938, Erfinder: Cortes F. Reed.
- Theodor Weyl (Begr.), Josef Houben (Hrsg.), Eugen Müller (Hrsg.): Methoden der organischen Chemie. IV/5a Photochemie. Thieme Verlag, Stuttgart 1975, ISBN 3-13-201904-6, S. 165–176.
- M. Le Blanc, K. Andrich: Photobromierung des Toluols. In: Zeitschrift für Elektrochemie und angewandte physikalische Chemie. 20.18‐19, 1914, S. 543–547, doi:10.1002/bbpc.19140201804.
- Klaus Schwetlick: Organikum. 23. Auflage. Wiley-VCH, Weinheim 2009, ISBN 978-3-527-32292-3, S. 206.
- Rudolf Bock: Gewinnung von Brom aus Meerwasser. In: Chemie Ingenieur Technik – CIT. 25, 1953, S. 245, doi:10.1002/cite.330250507.
- Hans-Friedrich Grützmacher, Jürgen Schmiegel: Dithia-diaza[n.2]metacyclophan-ene. In: Chemische Berichte. 122, 1989, S. 1929–1933, doi:10.1002/cber.19891221017.