Morbus Morquio
Morbus Morquio (Morquio-Krankheit) ist eine extrem seltene, angeborene Stoffwechselstörung aus der Gruppe der Mucopolysaccharidosen.
| Klassifikation nach ICD-10 | |
|---|---|
| E76.2 | Sonstige Mukopolysaccharidosen - Morquio-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Historisches
1929 wurde die seltene Krankheit erstmals von dem uruguayischen Kinderarzt Luis Morquió (1867–1935, aus Montevideo) beschrieben, und später nach ihm benannt. Gleichzeitig beschrieb auch der britische Radiologe James Frederik Brailsford (1888–1961, aus Birmingham) die Erkrankung, auch die Benennung als Morquio-Brailsford-Syndrom ist gebräuchlich. Meist wird aber die Bezeichnung Mukopolysaccharidose Typ IV (MPS-Typ IV) verwandt.
Es wurden mehrere Tonfiguren aus der Tumaco-La Tolita-Kultur gefunden, die vor etwa 2.500 Jahren im Grenzgebiet zwischen Kolumbien und Ecuador bestand. Diese Figuren zeigten Kleinwuchs, einen schmalen Brustkorb mit Schulterhochstand, eine kurze Nase, einen kurzen Hals und typische weitere Gesichtsveränderungen, die von Forschern als Darstellungen des Morbus Morquio interpretiert wurden.[1]
Formen
Die Ursache hierfür ist ein defektes Protein, genauer ein Enzym. Man unterscheidet je nach Enzymdefekt zwischen:
- Morbus Morquio Typ A. Dieser häufigeren Form liegt ein Sulfatase-Mangel (N-Acetyl-Galaktosamin-6-Sulfat-Sulfatase-Mangel) zugrunde, auf Gen 16q24.3 (No. 253000 im "Online Mendelian Inheritance in Man/OMIM"-Index)
- Morbus Morquio Typ B. Der selteneren Form liegt ein Beta-Galaktosidase-Mangel zugrunde, auf Gen 3p21.33 (No. 253010 im "Online Mendelian Inheritance in Man/OMIM"-Index)
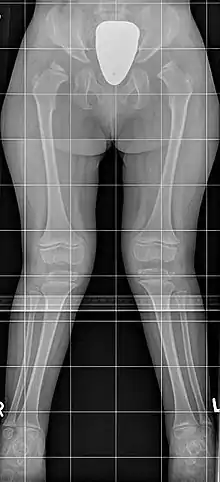
Erscheinung und Klinik
Die Schwere der Erkrankung ist sehr unterschiedlich, die Erkrankten des Typ B sind meist weniger stark betroffen. Es ist durchaus möglich, dass die Erkrankung wegen fehlender Beschwerden erst im Erwachsenenalter diagnostiziert wird, andere Patienten haben bereits als Kleinkinder deutliche körperliche Anzeichen. Typisch ist ein Kleinwuchs mit kurzem Hals und typischer Gesichtsform, sowie eine Hornhauttrübung und X-Beine, es besteht keine Intelligenzminderung und keine Vergrößerung von Milz oder Leber. Im Einzelnen finden sich:
- Minderwuchs: Die Endgröße beträgt selten mehr als 120 cm. Der Kleinwuchs ist durch die Verkürzung der langen Röhrenknochen dysproportioniert mit Betonung des Rumpfes. Dies fällt oft erst ab dem vierten Lebensjahr auf.
- Äußere Erscheinung: Typisch sind eine Kielbrust (Pectus carinatus; seltener eine Trichterbrust), eine Gelenk-Überbeweglichkeit (Hypermobilität) und X-Beine (Genua valga). Der Hals ist meist kurz. Das Gesicht zeigt typische Veränderungen, die an Wasserspeierfiguren (Gargoylen) an gotischen Kirchen erinnern und daher oft als Gargoylismus bezeichnet werden: Das Kinn ist vergrößert und vorstehend, das Gesicht vergröbert mit ausgeprägten Wangen bei großem Kopf (Makrozephalie).
- Wirbelsäule: Die Knochenveränderungen an der Wirbelsäule (spondyläre Dysplasie) sind so typisch, dass ein Röntgenbild häufig zur Diagnosestellung führt. Die Wirbelkörper sind flacher (Platyspondylie), vor allem die Wirbel am Übergang zwischen Brust- und Lendenwirbelsäule sind oft keilförmig mit daraus folgender Bildung eines Gibbus. Weiter besteht eine knöcherne Anlagestörung mit einem zu kleinen und nicht ausreichend fixierten Dens axis (des Zahnfortsatzes des zweiten Halswirbelkörpers), wodurch die Gefahr einer (atlantoaxialen) Instabilität besteht. Dies kann zu einer Spinalkanalstenose bis hin zur Querschnittsymptomatik führen. Eine Gefahr besteht besonders bei der endotrachealen Intubation im Rahmen einer Vollnarkose. Eine Rückenmarkkompression kann sich aber sowohl am Hals-Kopf-Übergang (atlantooccipital) als auch am Übergang zwischen Brust- und Lendenwirbelsäule (throrakolumbal) schleichend mit zunehmender Ermüdbarkeit und reduzierter Gehstrecke entwickeln. Am Becken besteht eine Hypoplasie des unteren Anteils des Os ilium sowie meist eine epiphysäre Dysplasie des Oberschenkelkopfes (Femurkopf-Epiphyse), die im Röntgenbild oft an einen Morbus Perthes erinnert. Es besteht meist eine Coxa valga. Die Mittelfußknochen (Ossa metatarsalia) sind körpernah zugespitzt, alle langen Röhrenknochen verkürzt.
- Augen: Hornhauttrübungen, die oft nur mit der Spaltlampe sichtbar sind.
- Zähne: meist Defekte im Zahnschmelz, die Zähne v. a. des Oberkiefers sind oft kleiner mit deutlicher Lückenbildung.
- Bauch: durch die Bindegewebsschwäche sind Nabel- und Leistenbrüche sehr häufig, die häufig operativ versorgt werden müssen
Genetik
Der Erbgang ist autosomal-rezessiv. Bei Typ A beschrieb Matalon 1974 einen Defekt einer 6-Sulfatase, DiFerrante konnte 1978 zeigen, dass es sich um die N-Acetyl-Galactosamin-6-Sulfat-Sulfatase (GALNS) handelt, die auf Vorschlag von Neufeld (1987) auch kurz als Galactose-6-Sulfatase bezeichnet wird. 1987 gelang Gibson die Reinigung und biochemische Beschreibung des Enzyms, das dann 1991 von der japanischen Arbeitsgruppe um Tomatsu geklont und sequenziert wurde. Dabei zeigte sich eine Sequenz von 552 Aminosäuren als Präpeptid, von denen ein N-terminales 26 Aminosäuren umfassendes Signalpeptid abgespalten wird, so dass die aktive Form ein 496 Aminosäuren umfassendes Peptid mit zwei Asparaginase-Glykolysierungsstellen ist. Es besteht eine hohe Homologie zu anderen Sulfatasen. Die Arbeitsgruppe lokalisierte das Protein auf Chromosom 16q24.3. Das dortige GALNS-Gen wurde von Nakashima 1994 näher analysiert. Es ist rund 50.000 Nukleotidbasen (50 kb) lang und umfasst 14 Exons. Analysen der Mutationen zeigten eine enorme Variabilität, Tomatsu summierte 2005 insgesamt 148 bekannte Mutationen auf dem GALNS-Gen. Diese große genetische Heterogenität ist für die ausgeprägte klinische Variabilität verantwortlich. Nelson schlug 1988 für Typ A eine Unterteilung in die klinischen Ausprägungen schwer-klassisch, intermediär und leicht vor.
Durch den Defekt eines Enzyms im Abbau der körpereigenen Glykosaminoglykane, die früher als Mucopolysaccharide bezeichnet wurden und die vorwiegend Gerüstproteine darstellen, fallen übermäßig Spaltprodukte an, die nicht weiter verarbeitet werden können. Diese werden daher zunächst in Lysosomen gespeichert und daher die Mucopolysaccharidosen zu den lysosomalen Speicherkrankheiten gezählt. Teilweise werden Zwischenprodukte auch vermehrt ausgeschieden, dies ist beim Morbus Morquio Typ A das Keratansulfat im Urin, bei Typ B Chondroitin-6-Sulfat. Vor allem werden die Spaltprodukte aber in charakteristischen Organen gespeichert – meist Skelettsystem, Bindegewebe, Augenabschnitte, Leber und Milz – und führen dort dann zu Funktionsstörungen. Anders als bei den anderen Mucopolysaccharidosen findet beim Morbus Morquio die Speicherung ausschließlich in Bindegewebezellen (v. a. Fibroblasten) statt, und weder im Zentralnervensystem noch in Leber oder Milz. Daher haben die Betroffenen eine normale Intelligenz und keine Vergrößerung der Leber oder Milz.[2] Seit 2014 ist eine spezifische Enzymersatztherapie für Morbus Morquio A mit Elosufase alfa möglich. Die ersten klinischen Studien sind vielversprechend.[3]
Eine pränatale Diagnostik mittels Chorionzottenbiopsie oder Amniozentese ist möglich.
Literatur
- Pressedienst der Johannes-Gutenberg-Universität, Klinikum Mainz
- Gesellschaft für MukoPolySaccharidosen
- Online Mendelian Inheritance in Man (OMIM). Johns Hopkins University:
- U. N. Riede, H. E. Schaefer: Allgemeine und spezielle Pathologie. 3. Auflage. Georg-Thieme-Verlag, Stuttgart 1993, ISBN 3-13-683303-1.
- B. Leiber: Die klinischen Syndrome. Urban und Schwarzenberg, München 1996, ISBN 3-541-01718-X.
Weblinks
- mps-ev.de (Deutsche Mukopolysaccharidose-Selbsthilfegruppe)
Einzelnachweise
- J. E. Bernal, I. Briceno: Genetic and other diseases in the pottery of Tumaco-La Tolita culture in Colombia-Ecuador. In: Clin. Genet. 2006; 70, S. 188–191.
- Monatsschrift Kinderheilkunde, Volume 155, Number 7, M.Beck: Lysosomal storage disorders.
- 1. Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. April 2014;74(6):713–82. Jones SA, Bialer M, Parini R, Martin K, Wang H, Yang K, u. a. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 years. Pediatr Res. 2. September 2015.