Fibröse Dysplasie
Die Fibröse Dysplasie (FD) ist eine angeborene Störung der Ossifikation. Durch die minderwertige Substantia compacta ist der Knochen aufgetrieben, verformt und bruchgefährdet. Eine andere Bezeichnung der (polyostotischen) FD ist Morbus Jaffé-Lichtenstein. Die Fibröse Dysplasie kommt auch im Rahmen eines McCune-Albright-Syndroms[1] (in Verbindung mit Pubertas praecox und Pigmentstörungen) vor.[2][3] Albrights Patient starb 1940 im Alter von 10 Jahren.[4] Vor der Schlesischen Gesellschaft für vaterländische Kultur berichtete Weil am 28. Juli 1922 über ein 9 Jahre altes Mädchen mit vorzeitiger Pubertät, brüchigen Knochen und dermaler Pigmentation.[5]
| Klassifikation nach ICD-10 | |
|---|---|
| M85.0 | Fibröse Dysplasie |
| ICD-10 online (WHO-Version 2019) | |
Natur
„A benign condition presumably developmental in nature, characterized by the presence of fibrous connective tissue with a characteristic whorled pattern and containing trabeculae of immature non-lamellar bone.“
Wichtig ist die Unterscheidung von polyostotischen und monostotischen Formen. Der typische Befall mehrerer oder aller Knochen ist eine gravierende und unheilbare Erkrankung. Dagegen ist der Einzelbefall meistens ein Zufallsbefund bei Erwachsenen; man kann abwarten oder kürettieren.
Symptome und Begleiterkrankungen
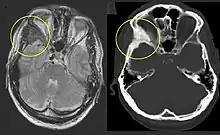
Die FD kann mit Myxomen der Skelettmuskulatur (Mazabraud-Syndrom) oder Funktionsstörungen von Herz, Leber, Bauchspeicheldrüse, Schilddrüse oder anderen Organen einhergehen.
Durch den verstärkten Knochenumbauprozess ist die FD oft mit einem erhöhten Wert der alkalischen Phosphatase im Blut sowie Hydroxyprolin bzw. Desoxypyridinolin im Urin verbunden. Kontrollen erlauben eine gewisse Verlaufsbeurteilung der FD.
Ursachen

Die FD wird durch eine nichtvererbbare Mutation des G-Proteins ausgelöst (im α-subunit). Die Mutation befindet sich im Gen GNAS (oder auch Gsα-Gen) des 20. Chromosoms. G-Proteine sind im Stoffwechsel der Zellen für die Signalweiterleitung extrem wichtig. Durch die Mutation kommt es zu einer Überaktivierung des Enzyms Adenylylcyclase, das die Katalyse von ATP zu cAMP steuert (das ist dann der eigentliche Signalübertragungsprozess). cAMP regelt z. B. die Herzfrequenz, Relaxation der glatten Muskulatur, die Wirkung von zahlreichen Hormonen und eben auch die Knochenzellen, die für den Aufbau von Knochen verantwortlich sind (Osteoblasten). Der Grund für diese Mutation ist noch unbekannt. Sie kann bereits im Föten-Stadium während der Schwangerschaft auftreten (dann treten die Symptome bereits im Kindes- und Jugendalter auf) oder auch erst später, nach der Geburt. Da die FD sehr stark mit Hormon- und anderen Zellstoffwechselprozessen verbunden ist, kann sie nach der Pubertät aufhören. An den eingetretenen Deformitäten ändert das nichts.
Der Gendefekt betrifft nur Körperzellen, nicht die Keimzellen (das erste nennt man dann somatische Mutation, dagegen bei Keimzellen gametische Mutation). Das heißt, der Gendefekt kann nicht an die Nachkommen weitergegeben werden. Findet die auslösende Mutation während der frühen embryonalen Entwicklung in der Zellmasse statt, kommt es vermutlich zum McCune-Albright-Syndrom; Mutation zu einem späteren Zeitpunkt der embryonalen Entwicklung löst vermutlich die polyostotische FD aus. Und eine Mutation nach der Geburt (im Kindes- oder gar Erwachsenenalter) wird für die monostotische FD verantwortlich gemacht. Je nachdem, wo die Mutation in der Zellmasse während der embryonalen Entwicklung stattfindet, gibt es später in diesen Körperregionen die Krankheit.
Diagnose

Die polyostotische FD ist klinisch und röntgenologisch auf den ersten Blick zu erkennen. Die befallenen Knochenbereiche weisen ein charakteristisches, milchglasartiges und unscharf begrenztes Erscheinungsbild im Röntgen oder CT auf. Eine Biopsie ist unnötig und erhöht die Bruchgefahr.
Behandlung
Bisphosphonate (Risedronate, Zoledronate) hemmen die Osteoklasten, behandeln aber wie die Operationen nicht die kausale Ursache der Erkrankung. Eingriffe am Achsskelett, wie Achskorrekturen und Stabilisierungen mit Marknägeln bergen einige Risiken. Bei Befall des Schädelknochens kann die Schutzfunktion desselben beeinträchtigt sein. Eine neurochirurgische Operation mit Exzision der Läsion im Gesunden und anschließender Rekonstruktion mit Kunstmaterialien (Titan oder Kunstmaterial wie PEEK, PMMA) kann entgegen den Eingriffen am Achsskelett ein risikoarmer und erfolgversprechender Therapieansatz sein.
Fibröse Dysplasie beim Neandertaler
An einem Abri am Hušnjakovo Berg in Krapina, Kroatien wurde bei einem vor über 120.000 Jahren verstorbenen Neandertaler aus dem Zeitalter des Moustérien ein Fragment einer linken Rippe mit Fibröser Dysplasie gefunden. Vermutlich handelt es sich um eine Rippe aus dem Bereich des 3. bis 6. Rippenbogens.[6]
Siehe auch
Literatur
- R. D. Chapurlat, P. J. Meunier: Fibrous dysplasia of bone. In: Best Practice & Research Clinical Rheumatology. Band 14, Nr. 2, 2000, S. 385–398, doi:10.1053/berh.1999.0071.
- R. Chapurlat: Current pharmacological treatment for fibrous dysplasia and perspectives for the future. In: Joint Bone Spine. Band 72, Nr. 3, 2005, S. 196–198, doi:10.1016/j.jbspin.2004.08.001.
- M. T. Collins, P. Bianco: American Society of Bone and Mineral Research. 2003, Fibrous Dysplasia, S. 466–470.
- M. T. Collins: McCune-Albright Syndrome. In: Orpha.net. 2010 (Zusammenfassung).
- R. Döhler, S. Hughes: Fibrous dysplasia of bone and the Weil-Albright syndrome. A study of thirteen patients with special reference to the orthopaedic treatment. In: International Orthopaedics. Band 10, 1986, S. 53–62.
- R. Döhler, W. A. Souter, I. Beggs, G. D. Smith: Idiopathic hyperphosphatasia with dermal pigmentation: A twenty year follow-up. In: Journal of Bone and Joint Surgery (Br). 68-B (1986), S. 305–310.
- T. E. Hullar, L. R. Lustig: Paget's disease and fibrous dysplasia. In: Otolaryngologic Clinics of North America. Band 36, Nr. 4, 2003, S. 707–732.
- M. H. Kelly, B. Brillante, H. Kushner, P. Gehron Robey, M. T. Collins: Physical function is impaired but quality of life preserved in patients with fibrous dysplasia of bone. In: Bone. Band 37, Nr. 3, 2005, S. 338–394, doi:10.1016/j.bone.2005.04.026.
- A. I. Leet, C. Chebli, H. Kushner, C. C. Chen, M. H. Kelly, B. A. Brillante, P. G. Robey, P. Bianco, S. Wientroub, M. T. Collins: Fracture Incidence in Polyostotic Fibrous Dysplasia and the McCune-Albright Syndrome. In: Journal of Bone and Mineral Research. Band 19, Nr. 4, 2004, S. 571–577, doi:10.1359/JBMR.0301262.
- P. Mikosch: Die Knochenszintigraphie in der Diagnostik metabolischer Knochenerkrankungen. In: WMW Wiener Medizinische Wochenschrift. Band 154, Nr. 5–6, 2004, S. 119–126, doi:10.1007/s10354-004-0053-4.
- L. S. Weinstein, S. Yu, D. R. Warner, J. Liu: Endocrine Manifestations of Stimulatory G Protein α-Subunit Mutations and the Role of Genomic Imprinting. In: Endocrine Reviews. Band 22, Nr. 5, 2001, S. 675–705 (Abstract).
- S. Wallach: Paget's disease and fibrous dysplasia. In: Current Opinion in Rheumatology. Band 3, Nr. 3, 1991, S. 472–480, PMID 1883702.
Weblinks
- Selbsthilfe-Seite (Seite nicht mehr abrufbar, festgestellt im August 2017. Suche in Webarchiven)
- Fibröse Dysplasie (PathoWiki) (Seite nicht mehr abrufbar, festgestellt im August 2017. Suche in Webarchiven)
Anmerkungen
- McCune-Albright-Syndrom: eine auch das zentrale Nervensystem betreffende Erkrankung mit Vorhandensein von körperlicher Frühreife (verfrühter Pubertät) und fibröser Dysplasie an mehreren Knochen (mit unsymmetrischem Gesicht oder deformiertem Skelett), Bezirken von Hautpigmentation, die an Milchkaffeeflecken erinnern, und anderen endokrin bedingten Störungen. Siehe Pschyrembel Klinisches Wörterbuch. Begründet von Willibald Pschyrembel. Bearbeitet von der Wörterbuchredaktion des Verlags. 255. Auflage. De Gruyter, Berlin 1986; und: Lois Jovanovic, Genell J. Subak-Sharpe: Hormone. Das medizinische Handbuch für Frauen. (Originalausgabe: Hormones. The Woman’s Answerbook. Atheneum, New York 1987) Aus dem Amerikanischen von Margaret Auer, Kabel, Hamburg 1989, ISBN 3-8225-0100-X, S. 80 und 380.
- D. J. McCune, H. Bruch: Osteodystrophia fibrosa. Report of a case in which the condition was combined with precocious puberty, pathologic pigmentation of the skin and hyperthyroidism, with a review of the literature. In: American Journal of Diseases in Childhood. 54 (1937), S. 806–848.
- F. Albright, A. M. Butler, A. O. Hampton, P. Smith: Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. In: The New England Journal of Medicine. 216 (1937), S. 727–746.
- Obduktionsbericht von H. E. MacMahon: Albrights syndrome – thirty years later. In: Pathology Annual. 1971, ISBN 0-407-95954-8, S. 81–146.
- Abstract in Klinische Wochenschrift. 1 (1922), S. 2114–2115.
- Janet Monge, Morrie Kricun, Jakov Radovčić, Davorka Radovčić, Alan Mann, David W. Frayer: Fibrous Dysplasia in a 120,000+ Year Old Neandertal from Krapina, Croatia. In: PLoS ONE 8(6): e64539. doi:10.1371/journal.pone.0064539.