Lomitapid
Lomitapid ist der erste Vertreter der neuen Wirkstoffklasse („first-in-class“) von Inhibitoren des mikrosomalen Triglycerid-Transfer-Proteins (MTP). MTP kommt im Lumen des endoplasmatischen Retikulums vor und ist für die Bindung und den Transport einzelner Lipidmoleküle zwischen Membranen verantwortlich.
| Strukturformel | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
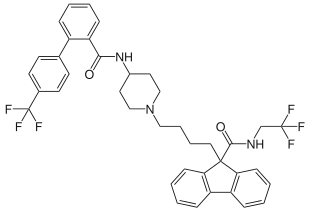 | |||||||||||||
| Allgemeines | |||||||||||||
| Freiname | Lomitapid | ||||||||||||
| Andere Namen |
| ||||||||||||
| Summenformel | C39H37F6N3O2 | ||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||
| |||||||||||||
| Arzneistoffangaben | |||||||||||||
| Wirkstoffklasse |
Sonstige Mittel, die den Lipidstoffwechsel beeinflussen | ||||||||||||
| Wirkmechanismus |
MTP-Hemmer | ||||||||||||
| Eigenschaften | |||||||||||||
| Molare Masse | 693,72 g·mol−1 | ||||||||||||
| Sicherheitshinweise | |||||||||||||
| |||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||||||||
Durch einen neuen Wirkansatz führt Lomitapid in Kombination mit anderen Maßnahmen zur zusätzlichen Senkung des im Rahmen der selten auftretenden homozygoten familiären Hypercholesterinämie stark erhöhten LDL-Cholesterinspiegels.
Anwendungsgebiete
Die US-amerikanische Zulassungsbehörde (FDA) hat Lomitapid in den USA im Dezember 2012 unter dem Namen Juxtapid, die Europäische Kommission im Juli 2013 als Lojuxta zugelassen zur Behandlung erwachsenen Patienten mit homozygoter familiärer Hypercholesterinämie (HoFH); die Gabe muss begleitend zu einer fettarmen Diät und anderen lipidsenkenden Arzneimitteln mit oder ohne Low-Density-Lipoprotein-Apherese (LDL-Apherese) erfolgen. Die Behandlung mit Lomitapid sollte von einem in der Behandlung von Lipidstörungen erfahrenen Arzt eingeleitet und überwacht werden.[2][3]
(→ zum Krankheitsbild siehe Homozygote familiäre Hypercholesterinämie)
HoFH ist eine seltene Krankheit, weswegen in den USA Lomitapid 2011 der Orphan-Drug-Status zuerkannt worden war.[4] Für Europa wurde dieser Status dagegen nicht zugebilligt.[5]
Die Diagnose HoFH sollte, wenn möglich, genetisch bestätigt werden. Andere Formen primärer Hyperlipoproteinämien sowie sekundäre Ursachen von Hypercholesterinämien (z. B. nephrotisches Syndrom oder Hypothyreose) müssen ausgeschlossen werden.
Um das Auftreten und die Schwere von gastrointestinalen Nebenwirkungen zu reduzieren, muss der Patient während der Anwendung den Fettanteil der täglichen Energiezufuhr konsequent unter 20 Prozent halten.
Die Sicherheit und Wirksamkeit von Lomitapid bei Kindern im Alter von unter 18 Jahren ist nicht erwiesen; daher wird die Anwendung dieses Arzneimittels bei Kindern nicht empfohlen.
Wirksamkeit
Die einarmige, unverblindete Zulassungsstudie[6] (Studienphase III) umfasste einen Zeitraum von 78 Behandlungswochen. Sechs Wochen vor Beginn der Behandlung wurden die Patienten angewiesen, ihre bereits bestehende lipidsenkende Therapie (Apherese in 18 Fällen angewandt) ohne Änderung beizubehalten und eine fettarme Diät einzuhalten (unter 20 Prozent der täglich zugeführten Energie von Fett stammend). Zudem wurde eine tägliche Supplementierung von Vitamin E und essentiellen Fettsäuren eingeleitet. Zu Studienbeginn hatten die Teilnehmer einen durchschnittlichen LDL-Cholesterinwert von 336 mg/dl. Bei Patienten, die das Verum erhielten, sank der LDL-Cholesterinwert im Mittel signifikant um 50 Prozent gegenüber den zu Beginn gemessenen Werten (von durchschnittlich 336 mg/dl auf durchschnittlich 166 mg/dl; p < 0,0001). In der sogenannten Intention-to-Treat (ITT)-Patientengruppe (29 Patienten) fiel der LDL-Cholesterinwert über 26 Wochen durchschnittlich um 40 Prozent (p < 0,001). 55 Prozent der ITT-Patienten erreichten während der 78 Wochen LDL-Cholesterinwerte unter 100 mg/dl, 31 Prozent unter 70 mg/dl – und damit die in der Europäischen Dyslipidämie-Leitlinie genannten Zielwerte für Patienten mit hohem und sehr hohem kardiovaskulärem Risiko. 65 Prozent der Studienpatienten konnten ihre lipidsenkende Begleittherapie reduzieren, einige konnten die LDL-Apherese stoppen oder die Intervalle strecken.
Es zeigten sich erhebliche Nebenwirkungen wie gastrointestinale Beschwerden bei den meisten Patienten bis hin zu einem Anstieg der Leberenzyme auf mehr als das Fünffache des oberen Normwertes. Die kernspintomographische Kontrolle zeigte, dass der hepatische Lipidgehalt von 1 % vor der Studie unter der Behandlung auf über 8 % nach 72 Wochen anstieg, da durch den Wirkstoff der Transport von Cholesterol von der Leber zu den Zellen blockiert ist.[7]
Wirkungsmechanismus
Lomitapid ist ein selektiver Inhibitor des mikrosomalen Triglycerid-Transfer-Proteins (MTP), eines intrazellulären Lipid-Transfer-Proteins, das im Lumen des endoplasmatischen Retikulums vorkommt und für die Bindung und den Transport einzelner Lipidmoleküle zwischen Membranen verantwortlich ist. Durch die selektive Hemmung des MTP durch Lomitapid kommt es zu einer verminderten Bildung von Lipidkomplexen – Very Low Density Lipoprotein (VLDL) in der Leber, Chylomikronen im Darm. Dadurch wird VLDL in vermindertem Maß von der Leber ins Blut abgegeben bzw. Chylomikronen werden weniger aus dem Darm aufgenommen. Hieraus resultiert eine konsekutive Senkung der Blutspiegel von VLD-Lipoprotein, Low Density Lipoprotein (LDL), LDL-Cholesterin, Chylomikronen und Apolipoprotein B (Apo B).[8]
Handelsnamen
- Lojuxta (EU), Juxtapid (CA, USA)
Zulassungsinhaber ist Aegerion Pharmaceuticals, ein Tochterunternehmen der Novelion Therapeutics. Für die Vermarktung in Europa wurde Lojuxta an Amryt Pharmaceuticals auslizenziert.
Literatur
- F.F.Smith, J.McKenney, LA.Bloedon, W.J.Sasiela, D.J.Rader: Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. In: Nature Clinical Practice. 5(8) 2008, S. 497–505.
Einzelnachweise
- Datenblatt Lomitapide bei Sigma-Aldrich, abgerufen am 19. Januar 2023 (PDF).
- Summary of the European public assessment report (EPAR) for Lojuxta, der EMA (engl.), abgerufen am 19. November 2015.
- Zusammenfassung des EPAR für die Öffentlichkeit der EMA (dt.), abgerufen am 19. November 2015.
- FDA Grants Orphan Drug Designation to Aegerion Pharmaceuticals' Drug Candidate, Lomitapide, for Treatment of Familial Chylomicronemia, 15. März 2011.
- Ärzte Zeitung online: Verzicht und Co.: Orphan Drugs oft ohne Orphan-Status, vom 13. November 2013, abgerufen am 11. Februar 2014.
- Marina Cuchel, Emma A Meagher u. a.: Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. In: The Lancet. 381, 2013, S. 40–46, doi:10.1016/S0140-6736(12)61731-0.
- Deutsches Ärzteblatt: Neuer Wirkstoff senkt extreme Cholesterinwerte, 2. November 2012, abgerufen am 2. Februar 2014.
- M Mahmood Hussain, Paul Rava, Meghan Walsh, Muhammad Rana, Jahangir Iqbal: Multiple functions of microsomal triglyceride transfer protein. In: Nutrition & Metabolism. 9, 2012, S. 14, doi:10.1186/1743-7075-9-14.