Sphingolipidose
Sphingolipidosen sind, bis auf den X-chromosomal vererbten Morbus Fabry, durch autosomal-rezessiv vererbte Gene ausgelöste Stoffwechselkrankheiten, die sich vorwiegend im zentralen Nervensystem manifestieren. Sie gehören zu der Gruppe der lysosomalen Speicherkrankheiten. Durch lysosomale Enzymdefekte oder -defizite, aber auch durch Defekte der Transport- oder Aktivatorproteine kommt es zu einer pathologischen intrazellulären Akkumulation von nicht weiter abbaubaren Sphingolipiden.
| Klassifikation nach ICD-10 | |
|---|---|
| E75 | Störungen des Sphingolipidstoffwechsels und sonstige Störungen der Lipidspeicherung |
| ICD-10 online (WHO-Version 2019) | |
Gemeinsame Symptome:
- motorische und geistige Retardierung
- Vergrößerung von Leber und Niere
- häufig kirschroter Makulafleck
Die metachromatische Leukodystrophie und Krabbe-Leukodystrophie sind demyelinisierende Erkrankungen, d. h. das Signalverhalten in kernspintomographischen Aufnahmen ist ähnlich wie bei anderen erworbenen demyelinisierenden Erkrankungen, wie z. B. Multiple Sklerose, Akute disseminierte Enzephalomyelitis, Balo-Sklerose, Neuromyelitis optica und Progressive multifokale Leukenzephalopathie.
Im Allgemeinen handelt es sich um schwere, noch in der Kindheit zum Tode führende Erkrankungen.
Varianten
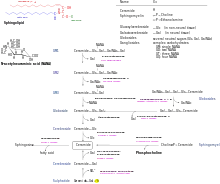
| Krankheit | Gespeicherte Substanz | Defektes Enzym |
|---|---|---|
| Niemann-Pick-Krankheit | Sphingomyelin | Sphingomyelinase |
| Morbus Gaucher | Glucocerebrosid | β-Glucosidase |
| Metachromatische Leukodystrophie | Sulfatide | Sulfatidase = Arylsulfatase A |
| Morbus Fabry | Globotriaosylceramid | α-Galactosidase |
| Tay-Sachs-Syndrom (amaurot. Idiotie, GM2-Gangliosidose) | Gangliosid GM2 | Hexosaminidase |
| Morbus Krabbe (globoidzellige Leukodystrophie) | Galactocerebrosid, Psychosin | Cerebrosid-β-Galactosidase |