Cushing-Syndrom
Das Cushing-Syndrom [] ist eine übermäßige Konzentration von Glucocorticoiden (Cortisol) im Blut, unter anderem mit erhöhtem Blutzuckerspiegel und bei chronischer Form typischen Körperformänderungen. Das Syndrom bzw. die Cushingsche Krankheit[1] wurde um 1943 nach dem US-amerikanischen Neurochirurgen Harvey Williams Cushing benannt, der es 1910 erstmals beschrieben hat.[2][3] Neben dem im Folgenden zunächst beschriebenen Cushing-Syndrom I wurden noch weitere Syndrome von Cushing beschrieben, die gelegentlich als Cushing-Syndrom II und III bezeichnet werden.
| Klassifikation nach ICD-10 | |
|---|---|
| E24 | Cushing-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
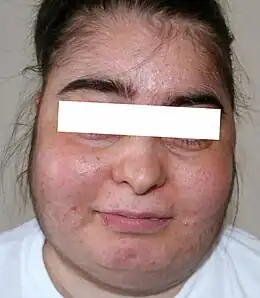
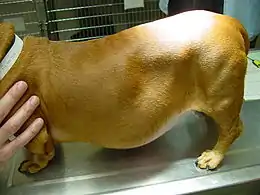
Cushing-Syndrom I
Beim Cushing-Syndrom handelt es sich um körperliche Veränderungen, die durch einen hohen Cortisolspiegel im Blut (Hypercortisolismus) verursacht werden. Dies ist gekennzeichnet durch einen erhöhten Blutzuckerspiegel und eine teilweise Unterdrückung des Immunsystems. Die Krankheit kann sich schleichend über Jahre entwickeln, wobei sich typischerweise am Körperstamm vermehrt Fettgewebe ansammelt und die Gliedmaßen durch Muskelschwund dünner werden.
Grundsätzlich unterscheidet man das endogene und das exogene Cushing-Syndrom. Letzteres ist wesentlich häufiger und wird von äußeren Einflüssen verursacht, insbesondere durch die längerfristige Einnahme von Glucocorticoiden (hauptsächlich Cortison).
Das endogene Cushing-Syndrom betrifft Frauen bis zu viermal häufiger als Männer und kann in allen Altersstufen auftreten. Dabei produziert die Nebennierenrinde, ohne dazu angeregt zu werden, zu viel Cortisol (Nebennierenrindenautonomie). Wird die Nebennierenrinde jedoch durch das Stimulationshormon ACTH angeregt, weil ein Tumor der Hypophyse vorliegt, wird die Erkrankung Morbus Cushing genannt.
Ursachen
Die Ursachen eines Cushing-Syndroms können sein:
- Zentrales Cushing-Syndrom (hypothalamisch-hypophysär): Erhöhte Produktion von ACTH (Adrenocorticotropes Hormon) im Hypophysenvorderlappen (Morbus Cushing, Cushingsche Erkrankung), seltener erhöhte Produktion von CRH im Hypothalamus mit anschließend vermehrter Kortikoidfreisetzung aus der Nebennierenrinde.
- Adrenales oder ACTH-unabhängiges Cushing-Syndrom: Gesteigerte Sekretion von Glukokortikoiden aus der Nebennierenrinde im Zuge von Neoplasien (Adenome oder Karzinome der Nebennierenrinde) mit nachfolgend unterdrückter ACTH-Ausschüttung aus dem Hypophysenvorderlappen.
- Ektopisches (paraneoplastisches) Cushing-Syndrom (auch ektopes ACTH-Syndrom genannt): Bildung von ACTH (oder sehr selten CRH) in ektopem Gewebe, meist im Rahmen eines kleinzelligen Bronchialkarzinoms.
- Durch Medikamente bedingtes Cushing-Syndrom: Ausbruch der Symptomatik nach regelmäßiger und längerer systemischer Einnahme hoher Dosen von ACTH oder Kortikoiden und anderen Steroidhormonen im Rahmen der Behandlung von Erkrankungen, etwa Autoimmunerkrankungen, Allergien oder Asthma bronchiale.
- Pseudo-Cushing-Syndrom (Cushingoid): Vorübergehende Cushing-Symptomatik, beispielsweise im Zuge von schweren Schädelverletzungen.
Symptomatik
Die Symptome des Cushing-Syndroms[4] sind durch die verstärkte hormonelle Wirkung der Kortikoide auf die Zielgewebe bedingt.
- (mäßige) Adipositas mit Bevorzugung des Stammes (Stammfettsucht)
- rundes, pausbackiges, gerötetes Gesicht (Vollmondgesicht)
- pralles Doppelkinn
- „Stiernacken“, auch „Büffelnacken“ und „Büffelhöcker“ genannt, bedingt durch Fettansammlung zwischen den Schultern
- Stoffwechsellage wie bei Diabetes mellitus mit Durst und häufigem Wasserlassen
- (geringe) arterielle Hypertonie
- oft Hirsutismus (übermäßige Behaarung) und Akne
- Gynäkomastie oder Pseudogynäkomastie
- Hypogonadismus, Libido- und Potenzminderung bei Männern
- Menstruationsstörungen (Oligo- und Amenorrhoe)
- Osteoporose
- Hautatrophie
- dünner werdende Haut und Bildung von rotviolettfarbenen breiten Gewebsstreifen (sogenannte Striae rubrae)
- leichte Ermüdbarkeit
- Verminderung der Muskelmasse (Muskelatrophie) mit schlankwirkenden Extremitäten (dünne Beine) mit proximaler Muskelschwäche der Extremitäten, auch Herzschwäche
- Suffusionen, vor allem an den Streckseiten der Vorderarme, durch erhöhte Kapillarfragilität
- positives Rumpel-Leede-Zeichen
- Plethora
- vermehrte Pigmentierung der Haut
- Wasseransammlungen im Gewebe (Ödeme)
- Bildung von Nierensteinen
- Rückenschmerzen
- erhöhte Infektanfälligkeit und langsames Heilen von Wunden, Pyodermien, Nagelmykosen
- plötzlich auftretende Gelenksschmerzen, die kommen und gehen
- endokrines Psychosyndrom (mit psychischen Wesensveränderungen)
Eine spezielle Form des Cushing-Syndroms mit ähnlicher Symptomatik ist das Achard-Thiers-Syndrom.
Diagnostik
Bei einem Verdacht auf das Cushing-Syndrom muss zunächst geklärt werden, ob kortisonhaltige Medikamente genommen wurden. Durch Blut-, Urin- und Speichel-Proben können entsprechende Hormonspiegel bestimmt werden. Vor allem mit dem Dexamethason-Suppressionstest und dem CRH-Test (s. u.) kann eine Störung der Ausschüttung von Nebennierenrindenhormonen erkannt werden. Zudem wird nach Tumoren wie Adenomen gesucht, die ein endogenes (im Körper selbst ausgelöstes) Cushing-Syndrom verursachen können. Mit Computer- oder Kernspinresonanztomographie wird nach Veränderungen der Hirnanhangsdrüse und der Nebennieren gesucht.
- Körperliche Untersuchung:
- Hautinspektion
- Blutdruckkontrolle
- Endokrinologisch:
- Dexamethason-Suppressionstest
- Kortisolausscheidung im 24-h-Urin
- Insulin-Hypoglykämietest
- CRH-Test
- Abklärung der Osteoporose
- Radiologisch:
- Darstellung der Nebennieren beziehungsweise der Hypophyse (Sonographie, CT, MRT)
Differenzierung hypophysär-adrenal-ektop
1. CRH-Test: 100 µg Corticotropin Releasing Hormon als intravenöse Bolusinjektion;
ACTH und Kortisol vor CRH und nach 30, 60, 90 und 120 Minuten messen.
| ACTH Spiegel | CRH-Test | |
|---|---|---|
| Normal | n | ACTH↑ Kortison↑ |
| Zentral | ↑ | ACTH↑ Kortison↑ |
| Adrenal | ↓ | ACTH↔ Kortison↔ |
| Ektop | ↑ | ACTH↔ Kortison↔ |
2. Liddle Test: 2 Tage 0,5 mg Dexamethason p.o. alle 6 Stunden, danach 2 Tage 2 mg Dexamethason alle 6 Stunden. Täglich Kortisolausscheidung im Harn sowie im Serumkortisol-Konzentration an Tag 3 und 5 um 8 Uhr morgens.
3. Sinus petrosus Sampling: Katheter in Sinus petrosus inferior beidseits.
ACTH, Kortisol und eventuell Prolaktin gleichzeitig aus Sinus petrosus links und rechts sowie aus peripherer Vene vor und 2, 5 und 10 Minuten nach CRH.
Cushing-Schwellendosis
In der Fachliteratur wird die Cushing-Schwellendosis mit der Wirkungsstärke von 7,5 mg Prednison oder 30–40 mg (bei Männern), 15–30 mg (bei Frauen) Cortisol pro Tag angegeben.
Bei Kindern beträgt die Cushing-Schwellendosis 6 mg / m² Körperoberfläche / Tag Prednisonäquivalent (Prednison ist ca. viermal so wirksam wie Cortisol). Dies beschreibt die tägliche Erhaltungsdosis, die gerade noch kein Cushing-Syndrom auslösen soll. Allerdings ist diese Angabe als grobe Richtlinie zu verstehen, da erhebliche inter-individuelle Unterschiede bestehen. Eine absolute Untergrenze, unter der die Glukokortikoidtherapie als sicher anzusehen ist, besteht nicht. Topisch (auf der Haut) angewandte Glukokortikoidpräparate verursachen mit höchster Wahrscheinlichkeit kein Cushing-Syndrom, da sie nur zu sehr geringem Teil die Hautbarriere durchdringen. Dennoch wird empfohlen, die Therapie auf maximal 20 % der Körperoberfläche zu beschränken und zurückhaltend bei der Anwendung im Gesicht oder am Auge zu sein.
Therapie
Die Therapie richtet sich nach der Ursache des Cushing-Syndroms: Adenome der Hypophyse oder der Nebennieren werden operativ entfernt; die Therapie der Wahl bei einer Nebennierenrinden-Hyperplasie ist die Adrenalektomie und eine sich anschließende lebenslange Hormonsubstitution zur Vermeidung der Ausbildung eines Morbus Addison. Als Komplikation kann sich danach ein Nelson-Tumor entwickeln.
Die EU-Kommission hat im Januar 2020 Osilodrostat (Handelsname: Isturisa, Hersteller: Novartis) als orale Behandlung von erwachsenen Patienten mit endogenem Cushing-Syndrom zugelassen. Osilodrostat ist Hemmstoff der körpereigenen Cortisol-Biosynthese.[5]
Cushing-Syndrom II
Das Cushing-Syndrom II ist eine seltene Erkrankung, bei der ein Tumor im Bereich von Pons oder Cerebellum gleichseitige Schädigungen der Hirnnerven VI, VII und VIII hervorruft. Ausfallerscheinungen des Kleinhirns und Hirndruckzeichen sind im Rahmen dieser Erkrankung ebenfalls beschrieben. Die aktuelle Bezeichnung lautet Kleinhirnbrücken-Symptomatik.
Der Erstbeschrieb erfolgte 1917 durch Harvey Williams Cushing.[6]
Diagnostische Kriterien sind:[7]
- Gangstörung, eventuell Tinnitus
- Gleichgewichtsstörung
- später Fazialisparese, Ausfall des Trigeminus
Cushing-Syndrom bei Tieren
In der Veterinärmedizin kommt ein Cushing-Syndrom vor allem beim Pferd (Equines Cushing-Syndrom), beim Hund (Canines Cushing-Syndrom) und selten bei der Katze (Felines Cushing-Syndrom) vor. Typische Laborveränderungen sind ein Stressleukogramm, erhöhte Leberwerte (ALT und ALP), ein erhöhter Blutzuckerspiegel, hohe Cholesterinwerte und eine geringes spezifisches Gewicht des Urins. Die Therapie der Erkrankung wird meist mit Medikamenten vorgenommen, die die Cortisolproduktion in der Nebennierenrinde senken. Mittel der Wahl für den Hund ist hierbei Trilostan.
Siehe auch
Weblinks
- The European Register on Cushing’s Syndrome
- Patienteninformation (PDF) [archiviert]
- Was ist ein Cushing-Syndrom? Internisten im Netz
- Folge 10 von "Abenteuer Diagnose", NDR-Podcast zum Thema
Einzelnachweise
- Paul Diepgen, Heinz Goerke: Aschoff/Diepgen/Goerke: Kurze Übersichtstabelle zur Geschichte der Medizin. 7., neubearbeitete Auflage. Springer, Berlin/Göttingen/Heidelberg 1960, S. 59.
- H. Cushing: The Pituitary Body and Its Disorders: Clinical States Produced by Disorders of the Hypophysis Cerebri. An Amplification of the Harvey Lecture for December, 1910. J. B. Lippincott, Philadelphia / London 1912.
- Die Benennung nach Cushing wurde 1942/43 von dem US-amerikanischen Endokrinologen Fuller Albright vorgeschlagen. Siehe: F. Albright: Cushing’s syndrome. In: Harvey Lectures. Series 38, 1943, S. 123–186.
- Vgl. hierzu W. Siegenthaler, U. Kuhlmann: Hypertonie. In: Walter Siegenthaler (Hrsg.): Differentialdiagnose innerer Krankheiten. 16., neubearbeitete Auflage, Georg Thieme Verlag, Stuttgart/ New York 1988, Kapitel 14, hier: Kapitel 14.18–14.25: Cushing-Syndrom (Hyperkortisolismus).
- Zusammenfassung der Merkmale des Arzneimittels. (PDF; 487 kB) EMA, abgerufen am 12. Februar 2020
- H. W. Cushing: Tumors of the nervus acusticus and the syndrome of the cerebellopontine angle. Saunders, Philadelphia 1917.
- B. Leiber: Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Herausgegeben von G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger, 7. Auflage. Urban & Schwarzenberg 1990, ISBN 3-541-01727-9.