Kaudales Regressionssyndrom
Das kaudale Regressionssyndrom ist ein seltenes Fehlbildungssyndrom des unteren Rumpfes, insbesondere der unteren Wirbelsäule, also der Lendenwirbelsäule (LWS) und des Steißbeins (Sakrum) sowie die Hüftgelenke. Betroffen sind neben den Knochen auch das Becken und der Beckenboden mit Enddarm und Harn- und Geschlechtsapparat.[1][2][3][4][5] Das Syndrom kann auch zu den Neuralrohrdefekten gezählt werden.[6]
| Klassifikation nach ICD-10 | |
|---|---|
| Q89.8 | Sonstige näher bezeichnete angeborene Fehlbildungen |
| ICD-10 online (WHO-Version 2019) | |
Synonyme sind: Kaudale Regression; Hypoplasie-Syndrom, kaudales; Dysplasie, kaudale; Syndrom der kaudalen Regression
Als "Gegenteil" kann die Kaudale Duplikation angesehen werden.
Die Erstbeschreibung stammt aus dem Jahre 1852 durch Anton Friedrich Hohl.[7] Der Begriff "Sakralagenesie" wurde im Jahre 1957 von D. I. Williams und H. H. Nixon verwendet,[8] der Begriff "Kaudales Regressionssyndrom" wurde im Jahre 1959 durch den französischen Kinderchirurgen Bernard Duhamel geprägt.[9]
Vorkommen
Die Häufigkeit wird mit etwa 1: 25 000–60 000 Lebendgeburten angegeben.[5] Das Syndrom tritt bei eineiigen Zwillingen häufiger auf. Das männliche Geschlecht ist 2,7 mal häufiger betroffen. Bei Diabetes mellitus der Schwangeren kommt diese Fehlbildung etwa 200 mal häufiger auf als in der Normalbevölkerung.[2]
Ursache
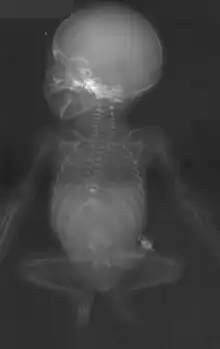
Als Ursache wird eine Entwicklungshemmung des Mesoderms vor der 4. (-12.) Schwangerschaftswoche angenommen.
Bei 16 % der Betroffenen bestand ein mütterlicher Diabetes.[2][1]
Es gibt Formen mit autosomal-dominanter oder rezessiver Vererbung mit Mutationen im VANGL1-Gen auf Chromosom 1 Genort p13.1.[10]
Beim Cerebroarthrodigital-Syndrom ist eine Sakralagenesie wesentliches Merkmal.[11]
Klinische Erscheinungen
Klinische Kriterien sind:[1][4][5]
- Verkürzung der unteren Rumpfhälfte wegen Agenesie/Hypogenesie der kaudalen Wirbelsäule, Kreuz & Steißbein fehlen meist ganz
- Dysplasie des Beckens
- Hypoplasie der unteren Extremität, oft Klumpfuß, Abduktionskontraktur der Hüftgelenke, Flexionskontraktur der Kniegelenke, Hypoplasie der Beinmuskulatur bis schlaffe Lähmung
- Anale Fehlbildungen, vor allem Analatresie.
- Urogenitale Fehlbildungen, Agenesie des Wolffschen oder Müllerschen Ganges, fehlende Hälfte des Uterus, Ovarialagenesie oder rektovaginale Fistel
- Kardiale Fehlbildungen
- oft Polyhydramnion/Oligohydramnion
- Sensomotorik ist ab dem letzten normalen Wirbelsegment gestört
Das Syndrom weist je nach Ausprägung ein breites Spektrum auf von nur partieller Agenesie der sacro-coccygealen Wirbelsäule bis zu schweren Deformitäten des Beckens mit Fusion der Beckenschaufeln, Anomalien der unteren Gliedmaßen (Flexion der Knie, Varusposition der Füße) und unterschiedlich schwer ausgeprägten motorischen und neurologischen Ausfällen (spontane motorische Aktivität und verringerte tiefe Sehnenreflexe der unteren Extremitäten).
In absteigender Häufigkeit findet sich eine Aplasie kaudal von SWK1, LWK1-5, bei BWK11 oder 12, schließlich bei BWK 9.[12]
Je nach Höhe der Defekte kommt es zu neurologischen Ausfällen, die von Störung der Mastdarm- und Harnblasenmotorik
(Blasenentleerungsstörung) bis zur schlaffen Lähmung der unteren Extremitäten reichen können.
Häufig ist der Konus oder das distale Myelon (siehe Rückenmark) deformiert.[13]
Einteilung
Je nach Schwere der Entwicklungshemmung können unterschieden werden:[1]
- Sakralhypoplasie mit Fehlen einzelner Kokzyx- und Sakrumelemente
- Sakralagenesie
- Lumbalagenesie (Lumbosakrale Agenesie) eventuell mit weiteren Wirbelkörperfehlbildungen (Segmentationsstörungen).
Als schwere Form ist die Sirenomelie anzusehen.
Nach T. S. Renshaw erfolgt folgende Klassifikation:[14][5]
- Typ I: Einseitige Sakralagenesie mit nicht progredienter Lumbalskoliose, normalen Hüft- und Kniegelenken, mögliche Fußfehlstellung
- Typ II: Partielle Sakralagenesie mit symmetrischen Defekten, hypoplastischen oder normalen Sakralwirbeln und einer stabilen Verbindung zu den Beckenschaufeln, bei zusätzlichen Wirbelkörperfehlbildungen wie Halbwirbeln fortschreitende Skoliose möglich. Hüftluxationen kommen vor, Knie und Füße sind nicht betroffen.
- Typ III: Unterschiedlich ausgeprägte Lumbalagenesie mit lumbaler Verbindung zu den Beckenschaufeln, stabile Verbindung, Hüftluxation, Kniegelenkskontraktur und Fußfehlbildung benötigen orthopädische Versorgung, um Stehen und Laufen zu können
- Typ IV: Lumbalagenesie ohne (stabile) Verbindung zu den Beckenschaufeln mit dysproportioniertem Kleinwuchs (Thorax zum Becken zu groß), horizontale Lage des Anus. Fast immer entwickelt sich eine fortschreitende Kyphose und Skoliose. Ausgeprägte Flexions- und Abduktionskontraktur der Hüften, Beugekontraktur der Knie mit Pterygiumbildung, Fußfehlbildung, fehlende Kontrolle über Harnblase und Enddarm.
Diagnostik
Die Diagnose kann bereits bei den Schwangerschaftsvorsorgeuntersuchungen mittels Ultraschall und/oder fetalem MRT erkannt werden.[2][15][16]
Nach der Geburt wird das Ausmaß der Malformationen durch Ultraschall und Kernspintomographie ermittelt, das Ausmaß der knöchernen Fehlbildung kann bereits auf einer Röntgen-Übersichtsaufnahme des Beckens mit LWS erfasst werden.
Differentialdiagnostik
Differentialdiagnostisch abzugrenzen sind:[1][2]
- Sirenomelie
- Familiäre kaudale Dysgenesie
- Currarino-Syndrom mit zusätzlicher Raumforderung vor dem Sakrum
- Kampomele Dysplasie
- Femur-Fibula-Ulna-Syndrom
Behandlung
Die Betreuung erfolgt in einem spezialisierten Zentrum mit multidisziplinärer Zusammenarbeit verschiedener Disziplinen. Je nach Ausmaß der Veränderungen sind mitunter zahlreiche operative Eingriffe notwendig.[2][5]
Aussichten
Die Prognose ist ungünstig. Überlebende Kinder sind geistig nicht beeinträchtigt.[2]
Einzelnachweise
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Kaudales Regressionssyndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Pschyrembel Online
- W. Schuster, D. Färber (Hrsg.): Kinderradiologie. Bildgebende Diagnostik. 2. Aufl., Springer 1996, ISBN 3-540-60224-0.
- emedicine.medscape
- A. van Baalen, J. M. Jacobs, K. Alfke et al: Kaudales Regressionssyndrom - kaudale Agenesie. In: Klinische Padiatrie. 2008 doi:10.1055/s-2007-993193.
- A. F. Hohl: Zur Pathologie des Beckens. Leipzig, 1852
- D. I. Williams, H. H. Nixon: Agenesis of the sacrum. In: Surgery, gynecology & obstetrics. Band 105, Nummer 1, Juli 1957, S. 84–88, PMID 13442908.
- B. Duhamel: Malformations ano-rectales et anomalies vertébrales. In: Archives françaises de pédiatrie, Band 16, S. 534, 1959.
- Caudal regression syndrome. In: Online Mendelian Inheritance in Man. (englisch)
- J. W. Spranger, A. Schinzel, T. Myers, J. Ryan, A. Giedion, J. M. Opitz: Cerebroarthrodigital syndrome: a newly recognized formal genesis syndrome in three patients with apparent arthromyodysplasia and sacral agenesis, brain malformation and digital hypoplasia. In: American journal of medical genetics. Band 5, Nummer 1, 1980, S. 13–24, doi:10.1002/ajmg.1320050104, PMID 7395897.
- J. P. Kuhn, T. L. Slovis, J. O. Haller (Hrsg.): Caffey’s Pediatric Diagnostic Imaging. 10. Auflage. Band I, Mosby 2004, ISBN 0-323-01109-8, S. 743f.
- J. P. Kuhn, T. L. Slovis, J. O. Haller (Hrsg.): Caffey’s Pediatric Diagnostic Imaging. 10. Auflage. Band I, Mosby 2004, ISBN 0-323-01109-8, S. 313.
- T. S. Renshaw: Sacral agenesis. In: The Journal of bone and joint surgery. American volume. Band 60, Nummer 3, April 1978, S. 373–383, PMID 649642.
- Y. Zheng, L. Li, L. Wang, C. Zhang: The clinical value of prenatal ultrasound in the diagnosis of caudal regression syndrome. In: American Journal of Translational Research. Band 15, Nummer 3, 2023, S. 1982–1989, PMID 37056862, PMC 1008693 (freier Volltext).
- M. H. Mahmoud, T. Asghar, T. H. Elkammash, A. M. Housseini, A. A. Gad: Fetal Magnetic Resonance Imaging in Association With Antenatal Ultrasound in the Diagnosis of Caudal Dysgenesis: Report of Two Cases. In: Cureus. Band 15, Nummer 2, Februar 2023, S. e35485, doi:10.7759/cureus.35485, PMID 36999114, PMC 1004564 (freier Volltext).
Literatur
- U. Purbasari, H. Nazar, F. Miraj, D. Aprilia, W. Widiani, M. Suprihatin, A. N. Eureka: Caudal regression syndrome from radiology and clinical perspective: A case series and a proposed new integrated diagnostic algorithm. In: Radiology case reports. Band 18, Nummer 7, Juli 2023, S. 2478–2486, doi:10.1016/j.radcr.2023.04.015, PMID 37235076, PMC 1020638 (freier Volltext).
- Barbara Jasiewicz, Wojciech Kacki: Caudal Regression Syndrome—A Narrative Review: An Orthopedic Point of View. In: . 2023, Band 10, Nummer 3, S. 589 doi:10.3390/children10030589.
- N. Mwamanenge, H. Mariki, M. Mkony, K. P. Manji: Caudal regression syndrome without maternal diabetes mellitus. In: BMJ Case Reports. Band 16, Nummer 3, März 2023, S. , doi:10.1136/bcr-2022-253136, PMID 36958756, PMC 1004000 (freier Volltext).
- Michael Entezami, Matthias Albig, Adam Gasiorek-Wiens, Rolf Becker: 11.13 Kaudales Regressionssyndrom. In: Georg Thieme Verlag eBooks. 2002 doi:10.1055/b-0034-60418.