Hodgkin-Lymphom
Das Hodgkin-Lymphom (Synonyme sind Morbus Hodgkin, früher Lymphogranulomatose oder Lymphogranulom, auch Hodgkinsche Krankheit; englisch Hodgkin’s disease, abgekürzt HD, oder Hodgkin’s lymphoma, abgekürzt HL) ist ein bösartiger Tumor („malignes Granulom“) des Lymphsystems.
| Klassifikation nach ICD-10 | |
|---|---|
| C81.0 | Lymphozytenreiche Form |
| C81.1 | Nodulär-sklerosierende Form |
| C81.2 | Gemischtzellige Form |
| C81.3 | Lymphozytenarme Form |
| C81.7 | Sonstige Form |
| C81.9 | nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Die Erkrankung macht sich durch schmerzlose Schwellungen von Lymphknoten bemerkbar, begleitend können sogenannte B-Symptome, wie zum Beispiel der für diese Erkrankung fast pathognomonische Alkoholschmerz (Schmerz in Lymphknotenregionen bereits bei geringer Alkoholaufnahme), auftreten. Im mikroskopischen Gewebebild ist das Hodgkin-Lymphom (genannt auch Sternbergsche Krankheit) durch das Vorkommen einer besonderen Zellart (Sternberg-Reed-Zellen) gekennzeichnet, wodurch es sich von den Non-Hodgkin-Lymphomen abgrenzt. Patienten werden mit standardisierten Therapieschemata durch eine Kombination aus Chemotherapie und Bestrahlung behandelt. Die Heilungsaussichten sind vor allem bei Kindern gut bis sehr gut.
Die Krankheit wurde nach dem englischen Arzt Thomas Hodgkin (1798–1866) benannt, der sie 1832 zum ersten Mal beschrieb.
Epidemiologie
Die Häufigkeit der jährlichen Neuerkrankungen (Inzidenz) des Hodgkin-Lymphoms beträgt zwei bis vier Erkrankungen pro 100.000 Personen, das Verhältnis von Männern zu Frauen liegt bei 3:2. In den Industrieländern findet man zwei Krankheitsgipfel in der Altersverteilung, einen größeren im dritten und einen etwas kleineren im siebten Lebensjahrzehnt, während in Entwicklungsländern typischerweise der erste Krankheitsgipfel in das frühe Kindheitsalter verschoben ist. In den industrialisierten Ländern (Europa, Nordamerika) ist eine leicht rückläufige Neuerkrankungsrate zu beobachten.
Bei Kindern und Jugendlichen in Deutschland tritt das Hodgkin-Lymphom mit einer Häufigkeit von 0,7 Fällen pro 100.000 Kindern und Jugendlichen im Alter bis 14 Jahren auf.[1] Im Vergleich hierzu liegt die entsprechende weltweite Rate bei 0,6 Fällen pro 100.000. Das Verhältnis Jungen zu Mädchen ist dabei analog zum Erwachsenenalter. Das mittlere Erkrankungsalter liegt bei 12 Jahren und 6 Monaten, der Altersgipfel liegt in der Altersgruppe von 10 bis 14 Jahren. Kinder unter vier Jahren sind selten vom Morbus Hodgkin betroffen, dabei übersteigt die Anzahl der erkrankten Jungen in dieser Altersgruppe die der Mädchen um ein Vielfaches.
Ursache
Die Ätiologie (Ursache) des Hodgkin-Lymphoms ist noch nicht hinreichend geklärt. In der Vergangenheit wurden viele Krankheitsauslöser diskutiert. Eine Entstehung durch das onkogene (krebsauslösende) Epstein-Barr-Virus (EBV) wird vermutet, da das Risiko, einen Morbus Hodgkin zu entwickeln, nach einem vorausgegangenen Pfeiffer-Drüsenfieber (infektiöse Mononukleose), das durch das EBV verursacht wird, etwa dreifach erhöht ist.[2] Bei 50 Prozent der Erkrankten in den industrialisierten Ländern lässt sich das Epstein-Barr-Virus in den Lymphomzellen nachweisen, in Entwicklungsländern beträgt diese Rate über 90 Prozent. Umgekehrt infiziert sich nahezu jeder Mensch irgendwann im Laufe seines Lebens mit dem EBV, im 30. Lebensjahr liegt die Durchseuchungsrate bei über 95 Prozent.
Störungen des Immunsystems kommt bei der Entstehung des Morbus Hodgkin eine bedeutende Rolle zu. Im Rahmen des zunehmenden Einsatzes immunsuppressiver (immunsystemunterdrückender) Therapien – beispielsweise nach Transplantationen von Organen, Knochenmark oder Blutstammzellen – wird ein vermehrtes Auftreten des Morbus Hodgkin berichtet.[3][4]
Auch die Infektion mit dem HI-Virus birgt ein erhöhtes Risiko, ein Hodgkin-Lymphom zu entwickeln, ebenso wie eine erhöhte Exposition toxischer Substanzen, zum Beispiel aus Holzschutzmitteln.[2]
Ende 2005 wurden in verschiedenen Arbeiten molekulare Mechanismen zur Pathogenese vorgeschlagen. Mathas und Kollegen identifizierten eine Störung des Transkriptionsfaktors E2A als mögliche Ursache einer Fehldifferenzierung der B-Lymphozyten.[5] Eine andere Gruppe publizierte die Degradierung des Tumorsuppressorgens Rb durch das latente Antigen 3C des Epstein-Barr-Virus in verschiedenen Tumoren.[6]
Die exakte Ursache ist also immer noch unbekannt und kaum Gegenstand aktueller Forschung, da der Großteil der Forschung nicht auf die Ursachensuche, sondern auf Therapieoptimierung ausgerichtet ist. Eine mögliche Impfung gegen EBV, um diesen Faktor der Entstehung auszuschalten oder weitere Erkenntnisse zu seiner Bedeutung zu gewinnen, steckt in der Entwicklungsphase.[7]
Pathologie
_CD30_immunostain.jpg.webp) |
_mixed_cellulary_type.jpg.webp) |
| immunhistochemische Färbung (CD30) | HE-Färbung |
| Histologische Schnitte eines befallenen Lymphknoten (gemischtzellige Form). Die prominenten Zellen mit mehreren, hellen Kernen mit deutlichen Nukleoli sind Sternberg-Reed-Zellen. | |
Kennzeichnend für die histologische (feingewebliche) Diagnose eines Hodgkin-Lymphoms sind die einkernigen Hodgkin-Zellen sowie die mehrkernigen Sternberg-Reed-Riesenzellen, oft auch als Hodgkin-Reed-Sternberg-Zellen (HRS-Zellen) bezeichnet. Diese stammen von den B-Lymphozyten (weißen Blutzellen) aus den Keimzentren der Lymphknoten ab.[8] Sie sind die eigentlichen maligne (bösartig) wachsenden Zellen des Hodgkin-Lymphoms und vermehren sich monoklonal (von einer Zelle abstammend). Typisch für Sternberg-Reed-Zellen ist dabei die Größe der Zelle von über 20 µm mit mehreren hellen Kernen, die jeweils große, eosinophile Nucleoli enthalten. Sie machen jedoch nur etwa ein Prozent des Lymphoms aus, der Rest wird durch reaktive Zellbeteiligung von CD4-positiven Lymphozyten, Monozyten, eosinophilen Granulozyten sowie Fibroblasten gebildet, wodurch sich ein „buntes“ zytologisches Bild ergibt.[9]
Die WHO unterscheidet in ihrer Klassifikation vier histologische Typen des sogenannten klassischen Hodgkin-Lymphoms von einer weiteren Form, dem lymphozytenprädominanten Lymphom. Die klassische Form ist durch die immunohistochemisch nachweisbaren Oberflächenmerkmale CD30 sowie teilweise CD15 gekennzeichnet. Die vier unterschiedlichen Typen sind im Einzelnen:
- Nodulär-sklerosierende Form (60 bis 80 Prozent der Fälle)
Typisch für diese häufigste Form des Hodgkin-Lymphoms sind knotige Infiltrate und Kollagennarben. Die bei diesem Typ beobachteten Lakunärzellen mit großem, gelapptem Kern sind eine Unterart der HRS-Zellen. Betroffen sind häufig junge weibliche Patienten vor allem bei mediastinalem und supraklavikulärem Befall. - Gemischtzellige Form (15 Prozent der Fälle)
Bei über 50 Jahre alten Patienten ist dies die häufigste Form des Hodgkin-Lymphoms, wobei Männer häufiger als Frauen betroffen sind. Ein zervikales, aber auch ein abdominelles Vorkommen ist typisch. Histologisch zeigt sich ein buntes Bild mit vielen HRS-Zellen. - Lymphozytenreiche Form (drei bis vier Prozent der Fälle)
Diese Form tritt meistens als zervikaler oder axillärer Lymphknotenbefall auf und kommt gehäuft bei männlichen Patienten um das 30. Lebensjahr vor. Histologisch dominieren B-Lymphozyten. - Lymphozytenarme Form (ein bis zwei Prozent der Fälle)
Diese seltene Form ist typisch für Patienten im hohen Alter und manifestiert sich oft im Bauchraum (Abdomen). Im Zellbild zeigen sich wenige Lymphozyten und atypische HRS-Zellen mit Mitosen.
Bei den heutigen Therapiemöglichkeiten unterscheiden sich die einzelnen Formen des klassischen Hodgkin-Lymphoms kaum in der #Prognose.
Den klassischen Formen steht das lymphozytenprädominante Hodgkin-Lymphom (Abkürzung: LPHD, frühere Bezeichnung: noduläres Paragranulom) gegenüber, typischerweise CD30- und CD15-negativ, dafür positiv für den B-Zell-Marker CD20. Die eigentlichen Tumorzellen dieses Typs sind lymphozytische und/oder histiozytische Zellen (L&H-Zellen), die große Ähnlichkeit mit den Hodgkin- und Sternberg-Reed-Zellen des klassischen Hodgkin-Lymphoms aufweisen können. Eine Besonderheit sind Popcorn-Zellen, eine Variation der HRS-Zellen. Der klinische Verlauf ist bei einer nur geringen Tendenz zur Metastasierung so gut, dass in lokalisierten Stadien (IA) eine Strahlentherapie ohne eine zusätzliche Chemotherapie ausreichend ist.
Klinisches Bild
Das Hodgkin-Lymphom beginnt zumeist mit schmerzlosen, zu Paketen verbackenen Lymphknotenschwellungen, die bei 80 bis 90 Prozent der Patienten zum Zeitpunkt der Diagnose vorhanden sind. Sie treten vor allem am Hals (zervikal), unter der Achsel (axillär) oder in der Leistenregion (inguinal) auf, jedoch auch im Mittelfell des Brustkorbs (mediastinal) und (bei der abdominellen Lymphogranulomatose) im Bauchraum (abdominal).
Begleitend kommt es zu unspezifischen Allgemeinsymptomen, der sogenannten B-Symptomatik. Darunter versteht man Fieber (gelegentlich als wellenförmiges Pel-Ebstein-Fieber), Nachtschweiß und eine (nicht anders erklärbare) Gewichtsabnahme von mehr als zehn Prozent innerhalb von sechs Monaten. Leistungsminderung und Juckreiz können ebenfalls bestehen. Selten ist eine Schmerzhaftigkeit der geschwollenen Lymphknoten nach Alkoholgenuss, dieser sogenannte Alkoholschmerz ist jedoch fast pathognomonisch für Hodgkin-Lymphome.[10] In manchen Fällen kann auch eine Leber- (Hepatomegalie) oder Milzvergrößerung (Splenomegalie) beobachtet werden.
In fortgeschrittenen Stadien mit Organbefall kann es zu Störungen des Nervensystems, des Hormonhaushaltes, des Urogenitaltraktes sowie zu Beschwerden bei Skelett- und Lungenbefall kommen. Eine Abschwächung des Immunsystems und infolgedessen gehäufte Infektionen, vor allem Tuberkulose, Pilz- und Virusinfektionen, sind möglich.
Hodgkin-Lymphome können auch durch paraneoplastische Syndrome in Erscheinung treten. Darunter versteht man Erkrankungen oder Symptomkomplexe, welche zumeist durch Autoimmunmechanismen verursacht werden, die wiederum auf einen bisweilen noch nicht diagnostizierten Morbus Hodgkin zurückzuführen sind. Mögliche paraneoplastische Syndrome sind Hauterkrankungen wie erworbene Ichthyosis[11] und Pemphigus[12] oder Erkrankungen des Nervensystems wie autonome, motorische und sensorische Neuropathien (Nervenschäden),[13] Encephalitis (Gehirnentzündung)[14] oder das so genannte Ophelia-Syndrom. bestehend aus Hippokampussklerose und Demenz.[15] Auch Autoimmunerkrankungen der Augen wie eine Entzündung der Lederhaut (Skleritis) kommen in diesem Zusammenhang vor.[16] Die paraneoplastischen Syndrome treten dabei oftmals vor der Ersterkrankung oder dem Rezidiv auf.
Diagnostik
Diagnosestellung
Die Verdachtsdiagnose basiert auf dem klinischen Bild, das durch Anamnese und Untersuchung erfasst wird. Hinweise geben auch Laborwerte: Als Entzündungszeichen sind oft die Blutsenkungsgeschwindigkeit und das C-reaktive Protein (CRP) erhöht. Typisch ist eine absolute Lymphopenie (Mangel an Lymphozyten, einer bestimmten Art weißer Blutkörperchen) (von bis zu < 1000/µl), und in einem Drittel der Fälle findet sich eine Eosinophilie. Im Labor zeigt sich weiterhin eventuell eine Anämie (Mangel an roten Blutkörperchen), eine Thrombopenie (Mangel an Blutplättchen) sowie eine LDH-Erhöhung. Mehr oder weniger unspezifisch sind ein erniedrigter Eisenwert und ein erhöhtes Ferritin.
Gesichert wird die Diagnose durch die histologische Untersuchung von Biopsien oder vollständig entnommenen verdächtigen Lymphknoten. Nach Möglichkeit soll die histologische Diagnose anhand eines vollständig entnommenen Lymphknotens durchgeführt werden[17]. Eine alleinige Feinnadelbiopsie gilt als unzureichend für die Diagnose[17].
Staging-Untersuchungen
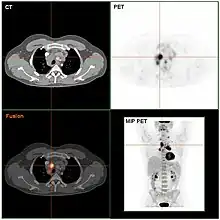
Das Ziel der folgenden klinischen Stadienbestimmung (klinisches Staging) ist es, alle Manifestationen zu erfassen und die Ausbreitung der Krankheit zu bestimmen. Das geschieht anhand der Befunde von Anamnese, Untersuchung, Laborwerten, Biopsien des Knochenmarks mit feingeweblicher Beurteilung sowie bildgebender Verfahren. Dazu gehören Röntgenbilder des Thorax in zwei Ebenen, Thorax-Computertomografie (CT), Sonografie (Ultraschall) und CT des Abdomens und eine Knochenmarkspunktion. Anstelle des CT kann bei bestimmten Patientengruppen mit Morbus Hodgkin auch die Magnetresonanztomographie (MRT) zum Einsatz kommen. Die Positronen-Emissions-Tomografie (PET) wird im Staging des Morbus Hodgkin zunehmend dann zusätzlich zur CT oder MRT eingesetzt, wenn die anderen vorgenannten bildgebenden Verfahren keinen ausreichend sicheren Aufschluss über einen Rückgang der Erkrankung unter Behandlung bieten. Ziel der PET-Untersuchungen soll sein, die Therapie noch besser nach der Erkrankungsaktivität zu steuern.
Die Methode des pathologischen Stagings mit Laparotomie (offener Bauchoperation) und Splenektomie (Milzentfernung) ist heute veraltet und wird nicht mehr durchgeführt.
Stadieneinteilung
Auf der Basis der Befunde des klinischen Stagings, aber unabhängig vom histologischen Typ wird das Hodgkin-Lymphom nach der Ann-Arbor-Klassifikation[18] (mit Modifikationen durch die Cotswolds-Konferenz 1989)[19] in vier Stadien eingeteilt:
| Stadium I | Befall einer einzigen Lymphknotenregion (IN) oder eines einzigen lokalisierten extranodalen Herdes (IE) |
| Stadium II | Befall von zwei oder mehr Lymphknotenregionen auf einer Seite des Zwerchfells (IIN) oder lokalisierte extranodale Herde und Befall einer oder mehrerer Lymphknotenregionen auf einer Seite des Zwerchfells (IIE) |
| Stadium III | Befall von zwei oder mehr Lymphknotenregionen auf beiden Seiten des Zwerchfells (IIIN) oder lokalisierte extranodale Herde auf beiden Seiten des Zwerchfells (IIIE) |
| Stadium IV | Verbreiteter (disseminierter) Befall eines oder mehrerer extralymphatischer Organe mit oder ohne Befall von Lymphknoten |
Zusätze:
A – ohne B-Symptome
B – mit B-Symptomen
E – extranodaler Befall (außerhalb von Lymphknoten)
S – Milzbefall (Spleen; englisch für Milz)
X – größere Tumor-Masse (Bulk oder bulky disease: Tumor > 10 cm maximaler Durchmesser bei Erwachsenen)
Bei Kindern und Jugendlichen gilt ein Befall des Knochens mit Zerstörung der Substanz (Compacta) oder ein Befall des Knochenmarks immer als Stadium IV, unabhängig von der Größe oder Anzahl der befallenen Lymphknotenstationen.
Therapie
Die Therapie des Hodgkin-Lymphoms basiert auf Chemotherapie und Bestrahlung. Die Therapie wird an das Stadium der Krankheit angepasst, wobei anhand des Ann-Arbor-Stadiums und vorhandener Risikofaktoren die drei Gruppen limitierte Stadien, intermediäre Stadien und fortgeschrittene Stadien eingeteilt werden (siehe Tabelle). Vor Beginn einer Chemotherapie sollte sichergestellt werden, dass Maßnahmen zur Sicherung der Fortpflanzungsfähigkeit des Patienten ergriffen wurden (zum Beispiel das Einfrieren von Spermien).
Therapiestudien
Die Deutsche Hodgkin-Studiengruppe (GHSG) erforscht seit 1978 die Therapiemöglichkeiten des Hodgkin-Lymphoms. Seitdem beteiligten sich über 14.000 Patienten an den Therapiestudien, an denen 400 Zentren beteiligt sind. Zurzeit (2014) ist die sechste Studiengeneration mit den Studien HD16 (limitierte Stadien) und HD18 (fortgeschrittene Stadien) sowie der Studie HD17 (intermediäre Stadien) aktuell. Unter anderem durch solche Studien gelang die erhebliche Prognoseverbesserung der letzten 20 Jahre, das Ziel der aktuellen Studien ist vor allem eine Verminderung der Nebenwirkungen der Therapie. Die GHSG veröffentlicht aufgrund dieser Studien Empfehlungen zur Therapie der verschiedenen Stadien, die aus einer Chemotherapie in Kombination mit einer Strahlentherapie besteht.
| Gruppe | Stadium / Risikofaktoren | Standardtherapie |
|---|---|---|
| limitierte Stadien limited disease |
Stadien I und II ohne Risikofaktoren | Studie: HD16
oder Chemotherapie: 2 × ABVD + Radiotherapie: 20 Gy involved field |
| intermediäre Stadien intermediate disease |
Stadien I und II mit Risikofaktoren (≥ 3 Lymphknoten Areale, Hohe BSG) Stadium I-IIA auch bei großem Mediastinaltumor (Bulk Tumor) oder extranodalem Befall | Studie HD17
oder Chemotherapie: 2 × BEACOPP eskaliert + 2 × ABVD + Radiotherapie: 30 Gy involved field |
| fortgeschrittene Stadien advanced disease |
Stadien IIB mit Bulk-Tumor, III und IV | Studie HD18 (18–60 Jahre)
oder Chemotherapie: 6 × BEACOPP eskaliert + Radiotherapie: 30 Gy involved field von PET-positivem Restgewebe ≥ 2,5 cm oder bei Patienten > 60 Jahre: 6,8 × ABVD + Radiotherapie: 30 Gy involved field von PET-positivem Restgewebe > 1,5 cm |
Als Risikofaktoren gelten:
- großer Mediastinaltumor (mehr als ein Drittel des Thoraxdurchmessers)
- Tumorwachstum außerhalb von Lymphknoten
- hohe Blutsenkungsgeschwindigkeit
- Befall von mehr als zwei Lymphknotenarealen
Bei Kindern und Jugendlichen wird in Deutschland die Diagnostik, Behandlung und Nachsorge (Nachbeobachtung) des Hodgkin-Lymphoms durch die multizentrische Therapieoptimierungsstudie HD-2003 der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) und der Deutschen Arbeitsgemeinschaft für Leukämieforschung und -Behandlung bei Kindern (DAL) durchgeführt. Alle kinderonkologischen Zentren in Deutschland behandeln nach dieser Studie. Der Studie HD-2003 angeschlossen sind Therapieoptimierungsstudien zur Behandlung von Rezidiven des Hodgkin-Lymphoms oder zur Behandlung des Hodgkin-Lymphoms in bestimmten Risikogruppen (angeborene oder erworbene Immundefekte). Die Behandlung von Kindern und Jugendlichen mit Hodgkin-Lymphom in Therapieoptimierungsstudien erfolgt seit 1978 (DAL-HD 78 Studie).[21] In anderen Staaten erfolgt die Behandlung ähnlich [in den USA beispielsweise gemäß den Therapieoptimierungsstudien durch die Children’s Oncology Group (COG)].
Bei Kindern sind Besonderheiten bei der Therapie zu berücksichtigen. Zum einen ist bei einem Großteil der Patienten das Wachstum nicht abgeschlossen. Dies führt zu besonderen Problemen in der Radiotherapie, insbesondere bei ausgedehntem Befall und somit ausgedehntem Bestrahlungsfeld, da die Radiotherapie selbst das Strahlenrisiko erhöht, d. h. spätere neue Krebserkrankungen befördert. Zum anderen wirken bestimmte Zytostatika toxisch auf die Spermienbildung. Auch weitere, wachstumsbedingte negative Effekte auf innere Organe erfordern angepasste Therapiepläne. Als wesentliche Unterscheidung zu Erwachsenen wird der Behandlungsplan auch nach dem Geschlecht ausgerichtet.
| Gruppe | Stadium | Geschlecht | Chemotherapie | Radiotherapie |
|---|---|---|---|---|
| limitierte Stadien limited disease |
IA, IB IIA |
Mädchen | 2 × OPPA (Woche 1–8) | wenn keine Vollremission nach Chemotherapie: involved field Radiotherapie (IF-RT) |
| Jungen | 2 × OEPA (Woche 1–8) | wenn keine Vollremission nach Chemotherapie: involved field Radiotherapie (IF-RT) | ||
| intermediäre Stadien intermediate disease |
IEA, IEB IIEA, IIB IIIA |
Mädchen | 2 × OPPA (Woche 1–8) + 2 × COPDIC (Woche 9–16) oder + 2 × COPP (Woche 9–16) | involved field Radiotherapie (IF-RT) |
| Jungen | 2 × OEPA (Woche 1–8) + 2 × COPDIC (Woche 9–16) oder + 2 × COPP (Woche 9–16) | involved field Radiotherapie (IF-RT) | ||
| fortgeschrittene Stadien advanced disease |
IIEB IIIEA, IIIEB, IIIB IV |
Mädchen | 2 × OPPA (Woche 1–8) + 4 × COPDIC (Woche 9–25) oder + 4 × COPP (Woche 9–25) | involved field Radiotherapie (IF-RT) |
| Jungen | 2 × OEPA (Woche 1–8) + 4 × COPDIC (Woche 9–25) oder + 4 × COPP (Woche 9–25) | involved field Radiotherapie (IF-RT) |
Chemotherapie
Die Chemotherapie wird als Poly- oder Kombinations-Chemotherapie durchgeführt. Eine solche soll nach Hudson und Donaldson folgende Eigenschaften aufweisen:
- Jedes eingesetzte Zytostatikum (Medikament) sollte eine Anti-Tumor-Aktivität (antineoplastische) Wirkung haben.
- Die eingesetzten Zytostatika (Medikamente) sollten sich hinsichtlich ihres Wirkungsmechanismus unterscheiden, um verschiedene Angriffspunkte gegenüber dem Tumor zu besitzen und eine Resistenzentwicklung zu verzögern.
- Die Toxizitäten der einzelnen Zytostatika sollten sich idealerweise nicht überlappen. Mindestens sollten die Toxizitäten der Zytostatika so sein, dass jedes einzelne Zytostatikum in seiner vollen Einzeldosis angewendet werden kann.
Nach diesen Grundsätzen werden verschiedene Zytostatika kombiniert. In internationalen Therapieprotokollen werden die Zytostatika dabei in festgelegten Dosierungen und Zyklen verabreicht. Abhängig vom Fortschritt der Erkrankung werden dabei verschiedene Protokolle und verschiedene Anzahl von Zykluswiederholungen eingesetzt. Die Schemata der Wahl sind das ABVD-Protokoll und das BEACOPP-Protokoll.
In Deutschland wird gemäß den Therapieempfehlungen der Deutschen Hodgkin-Studiengruppe ABVD bei limitierten (zwei Zyklen) und intermediären Stadien (vier Zyklen), BEACOPP bei fortgeschrittenen Stadien mit acht Zyklen in erhöhter (eskalierter) Dosis angewandt. Der Grund dafür ist, dass bei BEACOPP die Rezidivwahrscheinlichkeit etwas geringer ist, wobei jedoch Spätfolgen gegenüber ABVD etwas vermehrt vorkommen. Im Gegensatz dazu ist in den Vereinigten Staaten ABVD die Standardtherapie für alle Stadien, BEACOPP wird jedoch als Alternative ebenfalls angewandt.
Die Chemotherapie bei Kindern und Jugendlichen entspricht in ihren Grundlagen der Chemotherapie bei Erwachsenen: auch bei Kindern wird eine block- oder zyklusweise Poly-Chemotherapie durchgeführt. In Deutschland werden nachfolgende Kombinationen im Rahmen der Primärbehandlung des Morbus Hodgkin verwendet:
- OEPA (oder VEPA): Vincristin (Oncovin), Etoposid, Prednison, Adriamycin
- OPPA (oder VPPA): Vincristin (Oncovin), Procarbazin, Prednison, Adriamycin
- COPDIC: Cyclosphosphamid, Vincristin (Oncovin), Prednison, Dacarbazin
- COPP: wie bei Erwachsenen.
Im Gegensatz zum Erwachsenenalter bestimmt das Geschlecht bei der Chemotherapie im Kindesalter teilweise die verwendeten Kombinations-Chemotherapien: dem OPPA-Block wurde aufgrund der Toxizität auf die Spermienbildung das Procarbazin entnommen und durch das Etoposid ersetzt: so resultiert aus dem ursprünglichen OPPA-Block der OEPA-Block. Letzteren erhalten Jungen, ersteren die Mädchen, deren Eierstöcke deutlich unempfindlicher gegenüber Procarbazin sind als die Hoden der Jungen.
Chemoimmunkonjugat
Seit Ende 2012 ist in der Europäischen Union mit der Substanz Brentuximab Vedotin ein Antikörper-Wirkstoff-Konjugat (ADC) zur Behandlung des CD30+-Hodgkin-Lymphoms verfügbar. Das Arzneimittel ist zugelassen für die Therapie von erwachsenen Patienten mit einem rezidivierten oder refraktären CD30+-Hodgkin-Lymphom nach autologer Stammzelltransplantation oder nach mindestens zwei vorangegangenen Therapien, wenn eine autologe Stammzelltransplantation oder eine Kombinationschemotherapie nicht in Frage kommt.[22]
Ein immunphänotypisches Charakteristikum des klassischen Hodgkin-Lymphoms ist das CD30-Molekül.[23] Brentuximab Vedotin verknüpft einen anti-CD30 Antikörper über einen Aminosäure-Linker mit dem Zytostatikum Monomethyl-Auristatin E (MMAE).[24] Das Antikörper-Wirkstoff-Konjugat wurde bei mehrfach vorbehandelten und refraktären Hodgkin- sowie anderen CD30-positiven Lymphom-Patienten untersucht.[25][26][27]
Monoklonale Antikörper
Auch eine Therapie mit Nivolumab kann bei einem HL in Frage kommen. Die Evidenz ist sehr ungewiss bezüglich der Wirkung von Nivolumab auf das Gesamtüberleben, die Lebensqualität, das progressionsfreie Überleben, die Ansprechrate, die unerwünschten Ereignisse mit dem Grad 3 oder 4 und die schweren unerwünschten Ereignisse.[28]
Strahlentherapie
| Bestrahlungsfelder bei involved-field-Bestrahlung des Hodgkin-Lymphoms | |
|---|---|
| Befall | Bestrahlungsfelder |
| Stadium II Befall der Halslymphknoten rechts und der Lymphknoten im oberen Mediastinum | |
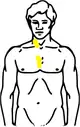 Hodgkin Stadium II (Schema) | 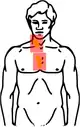 Hodgkin Stadium II (Schema) mit Bestrahlungsfeldern |
| Stadium III wie oben, zusätzlich: Lymphknotenbefall der linken Halsseite und Lymphknotenbefall unter dem Zwerchfell | |
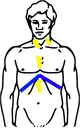 Hodgkin Stadium III (Schema) | 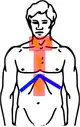 Hodgkin Stadium III (Schema) mit Bestrahlungsfeldern |
| Gelb: vom Hodgkin-Lymphom betroffene Areale Rot: Bestrahlungsfelder Blau: Zwerchfell | |
Bei einem nodulären Paragranulom (NLPHL) findet auch eine alleinige Bestrahlung ohne Chemotherapie statt.[2] Die Bestrahlung (Radiotherapie) erfolgt in der involved-field-Technik, worunter man eine Bestrahlung jedes klinisch manifesten Befalles unter Aussparung der angrenzenden Region versteht. Die empfohlene Gesamtdosis beträgt dabei je nach Stadium 20 oder 30 Gray (Gy), die in Einzeldosen von etwa 2 Gy pro Behandlungstag aufgeteilt wird. Weiterhin kommt nach Chemotherapie die konsolidierende Bestrahlung von ausgesuchten Tumorlokalisationen – wie Bulk-Regionen oder Resttumoren – in Frage.[2] In den Studien HD16 und HD17 wird bei negativem PET auf eine Bestrahlung nach Chemotherapie verzichtet, um die Spättoxizität der Behandlung zu mindern.[2]
Bei Kindern und Jugendlichen findet die Strahlentherapie ebenfalls Anwendung. Grundsätzlich folgt sie dabei den gleichen Prinzipien wie die Strahlentherapie bei Erwachsenen. Bestrahlt werden alle Regionen, die zum Diagnosezeitpunkt befallen waren (für die Risikogruppen intermediär und hoch beziehungsweise intermediate disease und extended disease). Dies bedeutet, dass die anfänglich betroffene Region bestrahlt wird, auch wenn sich unter Chemotherapie der Befall vollständig zurückgebildet hat. Die Bestrahlung erfolgt ebenfalls in der involved-field-Technik: das befallene Areal wird mit einem Sicherheitsabstand von 2 bis 3 cm bestrahlt. Die bei Kindern und Jugendlichen verwendete Gesamtdosis (Kumulativdosis) beträgt 20–30 Gy, die Aufteilung (Fraktionierung) wird mit Einzeldosen von 2 Gy pro Tag verabreicht.
Aufgrund des zumeist noch fortschreitenden Wachstums im Kindesalter ist die Bestrahlung größerer Regionen mit der involved-field-Technik bei ausgedehntem Befall (Stadium III oder IV nach Ann Arbor) nicht unproblematisch. Wenn oberhalb und unterhalb des Zwerchfells Lymphknotenregionen durch das Hodgkin-Lymphom betroffen sind, ergeben sich trotz der Begrenzung der Bestrahlungsfelder mittels der involved-field-Technik in der Summe große Bestrahlungsfelder. Diese enthalten oft Strukturen mit besonderer Bedeutung für das Wachstum, beispielsweise Wirbelsäule oder Schilddrüse.
Darüber hinaus ist der Zusammenhang zwischen Strahlentherapie des Hodgkin-Lymphoms und der Entstehung von Sekundärmalignomen (Zweitkrebserkrankung) gut beschrieben und mittlerweile etabliert. Die Häufigkeit oder Wahrscheinlichkeit, dass Zweitmalignome auftreten, hängt einerseits von der verwendeten Dosis und andererseits vom Bestrahlungsfeld und dessen Größe ab. Folgerichtig ist es Ziel von Therapieweiterentwicklungen, die Bestrahlung auf das unumgängliche Maß zu reduzieren. Eine Dosisabsenkung unter 20 Gy Gesamtdosis ist nicht sinnvoll, da bei einer solchen Dosis die Wirksamkeit der Bestrahlung gegen das Hodgkin-Lymphom eingeschränkt wird. Ein Ansatz verfolgt die Reduktion der Bestrahlungsfelder. Einerseits kann durch Intensivierung oder neue Chemotherapien die Notwendigkeit einer Bestrahlung vermindert werden. Zum anderen können unter Chemotherapie vollständig zurückgebildete befallene Regionen von der Bestrahlung ausgenommen werden. Um möglichst die therapeutischen Effektivität der Gesamtbehandlung nicht zu vermindern, werden zur Feststellung der Bestrahlungsnotwendigkeit Untersuchungsverfahren wie das PET eingesetzt.
Vitamin D
Ein Vitamin-D-Mangel verschlechtert die Prognose von Patienten mit einem Hodgkin-Lymphom.[29]
Supportivtherapie
Die Standardtherapie kann durch eine supportive Therapie unterstützt werden, die beispielsweise das Wohlbefinden der Patienten unterstützt. Eine Möglichkeit für eine Supportivtherapie könnte körperliche Betätigung sein. Die Evidenz erwies sich dabei als sehr ungewiss bezüglich des Effekts von körperlicher Betätigung auf Angst und schwere unerwünschte Ereignisse. Körperliche Betätigung verursache eventuell nur eine geringe oder keine Veränderung bezüglich der Mortalität, der Lebensqualität und der körperlichen Funktion.[30]
Therapieerfolgskontrolle (Re-Staging)
Um den Therapieerfolg von Chemotherapie und Strahlentherapie zu überprüfen, wird in regelmäßigen vorbestimmten Zeitintervallen eine erneute Diagnostik (Staging) durchgeführt. Diese Untersuchungen werden zusammenfassend als Re-Staging („Wieder-Einstufen“) bezeichnet. Beim Re-Staging kommen die gleichen Untersuchungsverfahren wie beim Staging im Rahmen der Erstdiagnose zum Einsatz. Die Ergebnisse des Re-Stagings werden mit den Ergebnissen des Stagings verglichen. Damit wird das Ansprechen auf die Therapie und das Ausmaß des Ansprechens festgestellt. In Abhängigkeit vom verwendeten Therapieprotokoll beziehungsweise der Therapieoptimierungsstudie sind Re-Staging-Untersuchungen nach zwei, vier oder sechs Chemotherapie-Zyklen sowie vor und nach der Strahlentherapie vorgesehen.
Eine Möglichkeit der Erfolgskontrolle der Therapie kann die Positronen-Emissions-Tomographie (PET) zum Beispiel nach dem zweiten Chemotherapiezyklus sein. Bei dem Vergleich von PET-negativen (= gute Prognose) und PET-positiven (= schlechte Prognose) Patienten erhielten Aldin und seine Mitarbeiter die folgenden Ergebnisse: Die Evidenz ist sehr ungewiss bezüglich der Wirkung des Effekts von negativen und positiven interimsmäßig durchgeführten PET-Untersuchungen auf das progressionsfreie Überleben. Negative interimsmäßige PET-Untersuchungen erzielen eventuell eine Erhöhung des progressionsfreien Überlebens im Vergleich zu positiven Untersuchungsergebnissen, wenn der angepasste Effekt gemessen wird. Negative interimsmäßige PET Resultate können zu einer ausgeprägten Erhöhung des Gesamtüberlebens im Vergleich zu positiven Resultaten führen. Dies gilt auch, wenn der angepasste Effekt gemessen wird.[31]
Nachsorge
Nach Therapieende/Remission erfolgt die Nachsorge meist alle drei Monate im ersten Jahr, alle sechs Monate ab dem zweiten Jahr und jährlich ab dem fünften Jahr.
Die Nachsorge besteht im Wesentlichen aus Sonografie des zuvor befallenen Bereiches sowie einer Blutuntersuchung. In längeren zeitlichen Abständen wird außerdem ein Röntgenbild des Thorax oder eine Computertomographie erstellt.
Rezidivtherapie
Tritt ein Rezidiv nach mehr als zwölf Monaten in vollständiger Remission auf, wird eine erneute Chemotherapie mit guter Chance für eine Langzeitremission durchgeführt. Ist die Phase der kompletten Remission kürzer oder ist die Remission nur unvollständig (primäres Therapieversagen), kann eine intensivierte Polychemotherapie (Salvage-Therapie) versucht werden. Alternativ wird eine myeloablative (knochenmarkselimierende) Hochdosis-Chemotherapie mit folgender Knochenmark- oder Stammzelltransplantation durchgeführt, wobei letztere auch autolog (durch Eigenspende während kompletter Remission) durchgeführt werden kann. Die Alternative ist eine allogene Blutstammzelltransplantation, wobei der Spender der Blutstammzellen nicht der Patient, sondern eine andere Person (verwandt oder nicht-verwandt) ist. Dieses Verfahren ist zum gegenwärtigen Zeitpunkt als experimentell einzustufen und wird im Rahmen von Studien international geprüft.[32] Bislang ist ein Nutzen allerdings nicht belegt.[33] Gleichsinniges gilt für Stammzelltransplantationen mit dosis- oder intensitätsreduzierter Konditionierungsbehandlung durch Chemotherapie oder Strahlentherapie (nicht-myeloablative Stammzelltransplantation; Mini-Transplant).[34]
Seit Ende 2012 ist in der Europäischen Union ein Chemoimmunkonjugat (Brentuximab Vedotin) zur Behandlung des rezidivierten oder refraktären Hodgkin-Lymphoms zugelassen.[22]
Behandlung von Nebenwirkungen
Neben den allseits bekannten Nebenwirkungen von Chemotherapie wie Haarausfall und Übelkeit, kann eine Chemotherapie oder eine Stammzelltransplantation auch zu Blutungen führen. Es hat sich herausgestellt, dass Thrombozytentransfusionen für Patienten mit einer Chemotherapie oder einer Stammzelltransplantation verschiedene Effekte auf die Anzahl der Patienten mit einem Blutungsereignis, die Anzahl der Tage mit einem Blutungsereignis, die Mortalität durch eine Blutung und die Anzahl der Thrombozytentransfusionen hatten, je nachdem in welcher Form sie verwendet worden sind (therapeutisch, abhängig von einem Schwellenwert, mit verschiedenen Dosierungen oder prophylaktisch).[35]
Im Jahr 2015 wurde die Verwendung von prophylaktischen Thrombozytentransfusionen zur Verhinderung von Blutungen evaluiert. Prophylaktische Thrombozytentransfusionen für Patienten mit einer Chemotherapie oder einer Stammzelltransplantation bei einem Schwellenwert von 10.000 verursachen eventuell nur eine geringe oder keine Veränderung bezüglich der Anzahl der Tage mit einem signifikanten Blutungsereignis pro Patient, der Anzahl der Teilnehmer mit einer Blutung der Stufe 3 oder 4 und der Zeit bis zur ersten Blutungsepisode. Prophylaktische Thrombozytentransfusionen verursachen eventuell eine geringe Verringerung der Anzahl von benötigten Transfusionen. Diese Transfusionen verursachen eventuell nur eine geringe Erhöhung bezüglich der Anzahl von Patienten mit einer Blutung. Prophylaktische Thrombozytentransfusionen bei einem Schwellenwert von 10.000 führen eventuell zu einer starken Erhöhung der Mortalität aus allen Gründen.[36]
Prognose
Gegen Ende des 20. Jahrhunderts ist es gelungen, die Überlebensraten deutlich zu steigern. Durch die stadienangepasste Therapie ist die Prognose mittlerweile auch für fortgeschrittene Stadien gut. Die Auswertung der dritten Studiengeneration der GHSG ergab eine Fünf-Jahres-Überlebensrate von über 90 Prozent auch für mittlere Stadien (HD8-Studie) und fortgeschrittene Stadien (HD9-Studie),[37][38] was durch die Zwischenergebnisse der vierten Studiengeneration gestützt wird.
Die Behandlungsergebnisse bei einem Rückfall oder Wiederauftreten des Hodgkin-Lymphoms hängen im Wesentlichen vom Zeitraum zwischen Abschluss der ersten Behandlung und Auftreten des Rückfalls ab. Wenn der Rückfall binnen drei bis zwölf Monate nach Ende der Erstbehandlung auftritt, ist die Prognose des Rückfalls mit nachfolgender Therapie schlechter als bei einem Rückfall, der mehr als zwölf Monate nach Ende der Erstbehandlung auftritt. Neben dem Zeitpunkt des Rückfalls sind auch die Ausmaße und Begleiterscheinungen des Rückfalls selbst von prognostischer Bedeutung. Ungünstig sind ein Rückfall mit Ausdehnung entsprechend Stadium III oder IV nach Ann Arbor, ein Hämoglobin-Wert von weniger als 10,5 g/dl bei Frauen und weniger als 12,0 g/dl bei Männern. Diese drei Kriterien oder Faktoren bestimmen nach Daten der Deutschen Hodgkin-Studiengruppe (GHSG; Internistische Onkologie) wesentlich die Prognose. Patienten, welche keines der drei Kriterien erfüllen, weisen eine rezidivfreie Vier-Jahre-Überlebensrate von 48 Prozent auf. Patienten, welche alle drei Kriterien erfüllen, weisen eine rezidivfreie Vier-Jahre-Überlebensrate von 17 Prozent auf.[39][40]
Patienten, die
- während oder nach der erstmaligen Behandlung ihres Hodgkin-Lymphoms nicht in Vollremission (komplettes Verschwinden der Krankheit) kommen oder
- unter laufender Therapie einen Progress (Fortschreiten) der Krankheit erfahren oder
- binnen drei Monaten nach Beendigung der Erstbehandlung einen Rückfall erleiden,
haben ebenfalls eine schlechte Prognose. Nach den Daten der GHSG beträgt die rezidivfreie Fünf-Jahre-Überlebensrate bei diesen Patienten 17 Prozent. Sofern eine Hochdosis-Chemotherapie durchgeführt wird, steigt die rezidivfreie Fünf-Jahre-Überlebensrate auf 42 Prozent. Allerdings erhalten nur 33 Prozent der Patienten mit den vorgenannten Kriterien eine Hochdosischemotherapie, da bei den verbleibenden 67 Prozent das Hodgkin-Lymphom rapide fortschreitet oder die Hochdosis-Chemotherapie mit einem extrem hohen Nebenwirkungsrisiko verbunden ist. Auch sind Patienten oftmals für eine geplante Hochdosis-Chemotherapie in einem nicht zureichenden Allgemeinzustand.[41]
Bei Kindern und Jugendlichen ist durch die Anwendung von multimodalen Therapieoptimierungsstudien die Prognose in den entwickelten Ländern exzellent. In Deutschland haben zwischen 1994 und 2003 96 Prozent aller 920 Fälle fünf Jahre überlebt, zehn Jahre nach Diagnosestellung lebten von den 920 Fällen noch 95 Prozent.[42]
Nach der Initialtherapie besteht eine gute Prognose. Die Langzeittoxizität von Radio- und Chemotherapie kann folgende Auswirkung haben:
Schädigung des Herzmuskels (durch Adriamycin und Bestrahlung) und der Lunge (durch Bleomycin und Bestrahlung), Schilddrüsenfunktionsstörungen sowie Störungen der Fruchtbarkeit wurden beobachtet.[43] Eine bedeutende Spätkomplikation ist die sekundäre Entwicklung anderer Krebsformen, insbesondere solider Tumoren, wie dem Mammakarzinom, dem Schilddrüsenkarzinom, dem Bronchialkarzinom[44], dem kolorektalen Karzinom und dem Magenkarzinom[45] oder hämatologischer Neoplasien wie der akuten myeloischen Leukämie,[46] eines myelodysplastischen Syndroms[47] oder eines Non-Hodgkin-Lymphoms. Die Erkrankungsrate an solchen Zweitneoplasien liegt etwa bei 15–20 Prozent in 20 Jahren[48], welche jedoch durch die Therapieform[49] und andere Faktoren[50] beeinflusst wird.[51][52][53]
Geschichtliche Aspekte
Der Morbus Hodgkin war nicht die erste Krebserkrankung, die entdeckt wurde, aber eine der ersten, für die wirksame Therapiemöglichkeiten entwickelt wurden. Wiederholte Verbesserungen der Therapie und deren klinische Überprüfung in Studien haben beim anfangs unheilbaren Morbus Hodgkin zu den heutigen Therapieerfolgen geführt.
Marcello Malpighi beschrieb 1666 als einer der Ersten in seiner Schrift De viscerum structura exercitatio anatomica wahrscheinlich ein Hodgkin-Lymphom.
Benannt wurde die Krankheit nach Thomas Hodgkin, der im Januar 1832 in seiner Arbeit On the morbid appearances of the Adsorbent Glands and Spleen verschiedene Fälle einer Krankheit, die das lymphatische System betrifft, beschrieb.[54]
1872 und 1878 veröffentlichten Langhans und Greenfield erstmals Arbeiten zu histopathologischen Aspekten der Krankheit, die Sternberg-Reed-Zellen wurden jedoch erst 1898 von Carl Sternberg[55] und 1902 von Dorothy Reed[56] unabhängig voneinander beschrieben. Benannt nach Sternberg erhielt das Hodgkin-Lymphom auch den Namen Sternbergsche Krankheit.[57]
Krumbhaar und Krumbhaar beobachteten 1919 erstmals, dass eine Senfgas-Vergiftung mit einer Leukopenie einhergeht.[58] 1931 führten Adair und Mitarbeiter die ersten experimentellen Untersuchungen über den Einsatz von Senfgas (Dichloroethylsulfid) bei Krebserkrankungen durch.[59] Im Zweiten Weltkrieg beobachtete man bei alliierten Soldaten, die nach dem Untergang des Munitionstransporters SS John Harvey (Bari, 2. Dezember 1943) Senfgas-Derivaten der N-Lost-Gruppe ausgesetzt waren,[60] eine Suppression von Knochenmark und Lymphsystem, was in den folgenden Jahren systematisch von verschiedenen Forschern wie Goodman (1946) untersucht wurde. Diese Beobachtungen und Untersuchungen mündeten in der Entwicklung von zunächst Mechlorethamin (Mustargen®), nachfolgend Cyclophosphamid (1959) und des darauf basierenden MOPP-Therapieschemas (1964), der ersten Kombinations-Polychemotherapie des Morbus Hodgkin.[61][62] Der erste publizierte Einsatz von Mechlorethamin in der Behandlung des Morbus Hodgkin bei Kindern erfolgte 1952.[63] In den folgenden Jahrzehnten wurde intensiv zu den Kombinationschemata geforscht und es wurden immer neue Therapien entwickelt, wie beispielsweise MOPP, ABVD, COPP und BEACOPP.
Im April 1971 wurden bei der Konferenz in Ann Arbor, USA, wichtige Definitionen zur Diagnose und Klassifikation (Ann-Arbor-Klassifikation) festgelegt. Die Deutsche Hodgkin-Studiengruppe erforscht seit 1978 die Effektivität verschiedener Therapien in großen, multizentrischen Studien und hat dadurch wesentlich zu den aktuellen Therapieempfehlungen und der damit verbundenen Prognoseverbesserung beigetragen.
1975 gelang Milstein und Köhler erstmals die Herstellung monoklonaler Antikörper, was als maßgebliche Grundlage für heutige Antikörpertherapien 1984 mit dem Nobelpreis für Medizin honoriert wurde. Auf dieser Basis wurde in den 1990er Jahren der Antikörper Rituximab für die Therapie der Non-Hodgkin-Lymphome eingeführt, 2002 wurde ein erfolgreicher Einsatz auch bei der lymphozyten-prädominanten Form des Morbus Hodgkin nachgewiesen. Eine Pilotstudie von Younes et al. zur Therapie beim klassischen Hodgkin-Lymphom läuft seit 2003.
Im Dezember 2005 veröffentlichten Mathas und Mitarbeiter[5] sowie Knight, Robertson und deren Mitarbeiter[6] verschiedene molekulare Mechanismen zur Entstehung der Krankheit, die vorher weitgehend unklar und lange Gegenstand intensiver Forschung gewesen waren.
Literatur
Lehrbücher
- P. Calabresi, B. A. Chabner: Antineoplastic Agents. In: A. G. Gilman, T. W. Rall, A. S. Nies, P. Taylor (Hrsg.): Goodman and Gilman’s the pharmacological basis of therapeutics. 8., internationale Ausgabe. McGraw-Hill Health Professions Division, New York / St. Louis / San Francisco 1992, ISBN 0-07-112621-X.
- B. A. Chabner, D. L. Longo (Hrsg.): Cancer Chemotherapy and Biotherapy: Principles and Practice. 2. Auflage. Lippincott Williams & Wilkins Publishers, Philadelphia / Baltimore / New York 1996, ISBN 0-397-51418-2.
- V. DeVita, S. Hellman, S. A. Rosenberg: Cancer. Principles and Practice of Oncology. 6. Auflage. Lippincott Williams & Wilkins Publishers, Philadelphia / Baltimore / New York 2000, ISBN 0-7817-2387-6.
- F. L. Greene, D. L. Page, I. D. Fleming, A. Fritz, C. M. Balch: AJCC Cancer Staging Handbook. 6. Auflage. Springer, New York / Berlin / Heidelberg 2002, ISBN 0-387-95271-3.
- Markus Sieber, Andrea Staratschek-Jox, Volker Diehl: Hodgkin-Lymphom. (PDF) In: Lehrbuch der Klinischen Onkologie (Springer). v. W. Hiddemann, C. Bartram, H. Huber, archiviert vom (nicht mehr online verfügbar) am 3. Dezember 2008; abgerufen am 20. August 2009.
- P. A. Pizzo, D. G. Poplack: Principles and Practice of Pediatric Oncology. 4. Auflage. Lippincott Williams & Wilkins Publishers, Philadelphia / Baltimore / New York 2001, ISBN 0-7817-2658-1.
- R. L. Souhami, I. Tannock, P. Hohenberger, J.-C. Horiot: Oxford Textbook of Oncology. 2. Auflage. Oxford University Press, Oxford 2002, ISBN 0-19-262926-3.
Leitlinien
- S3-Leitlinie Hodgkin-Lymphom, Diagnostik, Therapie und Nachsorge von erwachsenen Patienten der Deutschen Gesellschaft für Hämatologie und Onkologie (DGHO). In: AWMF online (Stand 2013)
- S1-Leitlinie Hodgkin-Lymphom der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). In: AWMF online (Stand 2007)
Weblinks
- Deutsche Hodgkin-Studiengruppe (GHSG)
- Kompetenznetz Maligne Lymphome
- Detaillierte Informationen zur historischen Entwicklung der Hodgkin-Forschung (englisch)
- Informationen zum Morbus Hodgkin bei Kindern und Jugendlichen der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH), Deutschland
- Hodgkin-Lymphom. (PDF; 1,36 MB) Ratgeber der Deutschen Krebshilfe (Die blauen Ratgeber 21)
Einzelnachweise
- Jahresbericht 2004 des Deutschen Kinderkrebsregisters (DKKR) der Universität Mainz. (PDF) Deutsches Kinderkrebsregister, archiviert vom (nicht mehr online verfügbar) am 28. September 2007; abgerufen am 19. März 2011.
- Herold: Innere Medizin: eine vorlesungsorientierte Darstellung: unter Berücksichtigung des Gegenstandskataloges für die Ärztliche Prüfung: mit ICD-10-Schlüssel im Text und Stichwortverzeichnis. Gerd Herold, Köln 2013, ISBN 978-3-9814660-2-7.
- A. Zambelli u. a.: Hodgkin’s disease as unusual presentation of post-transplant lymphoproliferative disorder after autologous hematopoietic cell transplantation for malignant glioma. In: BMC Cancer. Band 5, 23. August 2005, S. 109, PMID 16117828
- S. Caillard u. a.: Myeloma, Hodgkin disease, and lymphoid leukemia after renal transplantation: characteristics, risk factors and prognosis. In: Transplantation. Band 81, Nr. 6, 27. März 2006, S. 888–895. PMID 16570013
- S. Mathas u. a.: Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. In: Nature Immunology. Band 7, Nr. 2, 2006, S. 207–215, PMID 16369535
- J. S. Knight u. a.: Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. In: PNAS. Band 102, Nr. 51, 2005, S. 18562–18566, PMID 16352731, PMC 1317900 (freier Volltext)
- Henry H. Balfour Jr.: Progress, prospects, and problems in Epstein-Barr virus vaccine development. In: Current Opinion in Virology. Band 6, Juni 2014, ISSN 1879-6257, S. 1–5, doi:10.1016/j.coviro.2014.02.005.
- J. Cossman u. a.: Reed-Sternberg cell genome expression supports a B-cell lineage. In: Blood. Band 94, Nr. 2, 1999, S. 411–416, PMID 10397707.
- A. C. Feiler, H. Herbst, A. Marx: Lymphatisches System. In: W. Böcker, H. Denk, Ph. U. Heitz, H. Moch (Hrsg.): Pathologie. 4. Auflage. München 2008, S. 565–567.
- Kompetenznetz Maligne Lymphome. Abgerufen am 28. März 2014.
- E. Rizos u. a.: Acquired icthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. In: The Lancet Oncology. Band 3, Nr. 12, 2002, S. 727, PMID 12473513
- W. Tilakaratne und M. Dissanayake: Paraneoplastic pemphigus: a case report and review of literature. In: Oral Diseases. Band 11, Nr. 5, September 2005, S. 326–329. PMID 16120122
- B. C. Oh u. a.: A case of Hodgkin’s lymphoma associated with sensory neuropathy. In: J Korean Med Sci. Band 19, Nr. 1, Februar 2004, S. 130–133, PMID 14966355
- S. Kung u. a.: Delirium resulting from paraneoplastic limbic encephalitis caused by Hodgkin’s disease. In: Psychosomatics. Band 43, Nr. 6, November-Dezember 2002, S. 498–501, PMID 12444235.
- T. Shinohara u. a.: Pathology of pure hippocampal sclerosis in a patient with dementia and Hodgkin’s disease: the Ophelia syndrome. In: Neuropathology. Band 25, Nr. 4, Dezember 2005, S. 353–360, PMID 16382785.
- M. M. Thakker u. a.: Multifocal nodular episcleritis and scleritis with undiagnosed Hodgkin’s lymphoma. In: Ophthalmology. Band 110, Nr. 5, Mai 2003, S: 1057–1060, PMID 12750114.
- Prof. Dr. Nicole Skoetz, Prof. Dr. Andreas Engert, Nina Kreuzberger: S3-Leitlinie Diagnostik, Therapie und Nachsorge des Hodgkin Lymphoms bei erwachsenen Patienten. Leitlinienprogramm Onkologie der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF) , der Deutschen Krebsgesellschaft e.V. (DKG) und der Deutschen Krebshilfe., 1. Oktober 2022, S. 29, abgerufen am 19. Dezember 2023.
- F. L. Greene u. a.: AJCC Cancer Staging Handbook. 6. Auflage. Springer, New York/ Berlin/ Heidelberg 2002.
- T. Lister u. a.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. In: Journal of Clinical Oncology. 1989, Nr. 7, S. 1630.
- Therapie - GHSG - German Hodgkin Study Group. Abgerufen am 28. März 2014.
- G. Schellong: Cooperative therapy study HD 78 for Hodgkin’s disease in children and adolescents. In: Monatsschrift für Kinderheilkunde Band 127, Nr. 8, August 1979, S. 487–489, PMID 470955
- Summary of the European public assessment report (EPAR) for Adcetris
- YE Deutsch, T Tadmor, ER Podack, u. a. CD30: an important new target in hematologic malignancies. In: Leukemia & Lymphoma. Band 52, 2011, S. 1641–1654 PMID 21619423
- N. M. Okeley, J. B. Miyamoto, Zhang X. u. a. Intracellular activation of SGN-35, a potent anti-CD30 anti-body-drug conjugate. In: Clinical Cancer Research. Band 16, 2010, S. 888–897, PMID 20086002
- A. Rothe, S. Sasse, H. Goergen u. a.: Brentuximab vedotin for relapsed or refractory CD30+ hematologic malignancies: the German Hodgkin Study Group experience. In: Blood. Band 120, 2012, S. 1470–1472, PMID 22786877
- A. Younes, A. K. Gopal, S. E. Smith u. a.: Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients With Relapsed or Refractory Hodgkin’s Lymphoma. In: Journal of Clinical Oncology. Band 30, 2012, S. 2183–2189, PMID 22454421
- B. Pro, R. Advani, P. Brice u. a.: Brentuximab Vedotin (SGN-35) in Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. In: Journal of Clinical Oncology. Band 30, 2012, S. 2190–2196, PMID 22614995
- Marius Goldkuhle, Maria Dimaki, Gerald Gartlehner, Ina Monsef, Philipp Dahm: Nivolumab for adults with Hodgkin’s lymphoma (a rapid review using the software RobotReviewer). In: Cochrane Database of Systematic Reviews. 12. Juli 2018, doi:10.1002/14651858.CD012556.pub2 (wiley.com [abgerufen am 9. Juli 2020]).
- Sven Borchmann, Melita Cirillo u. a.: Pretreatment Vitamin D Deficiency Is Associated With Impaired Progression-Free and Overall Survival in Hodgkin Lymphoma. In: Journal of Clinical Oncology. 2019, doi:10.1200/JCO.19.00985.
- Linus Knips, Nils Bergenthal, Fiona Streckmann, Ina Monsef, Thomas Elter: Aerobic physical exercise for adult patients with haematological malignancies. In: Cochrane Database of Systematic Reviews. 31. Januar 2019, doi:10.1002/14651858.CD009075.pub3 (wiley.com [abgerufen am 9. Juli 2020]).
- Angela Aldin, Lisa Umlauff, Lise J. Estcourt, Gary Collins, Karel G. M. Moons: Interim PET-results for prognosis in adults with Hodgkin lymphoma: a systematic review and meta-analysis of prognostic factor studies. In: Cochrane Database of Systematic Reviews. 13. Januar 2020, doi:10.1002/14651858.CD012643.pub3 (wiley.com [abgerufen am 9. Juli 2020]).
- N. Schmitz, A. Sureda: The role of allogeneic stem-cell transplantation in Hodgkin’s disease. In: Eur J Haematol Suppl. Band 66, Juli 2005, S. 146–149, PMID 16007884
- Deutsches Ärzteblatt, 28. September 2010: Nutzen der allogenen Stammzelltransplantation bei Hodgkin nicht belegt (Memento vom 21. April 2015 im Internet Archive)
- I. Alvarez u. a.: Nonmyeloablative stem cell transplantation is an effective therapy for refractory or relapsed hodgkin lymphoma: results of a spanish prospective cooperative protocol. In: Biology of Blood and Marrow Transplantation. Band 12, Nr. 2, Februar 2006, S. 172–183, PMID 16443515
- Lise Estcourt, Simon Stanworth, Carolyn Doree, Sally Hopewell, Michael F. Murphy: Prophylactic platelet transfusion for prevention of bleeding in patients with haematological disorders after chemotherapy and stem cell transplantation. In: Cochrane Database of Systematic Reviews. 16. Mai 2012, doi:10.1002/14651858.CD004269.pub3 (wiley.com [abgerufen am 9. Juli 2020]).
- Lise J. Estcourt, Simon J. Stanworth, Carolyn Doree, Sally Hopewell, Marialena Trivella: Comparison of different platelet count thresholds to guide administration of prophylactic platelet transfusion for preventing bleeding in people with haematological disorders after myelosuppressive chemotherapy or stem cell transplantation. In: Cochrane Database of Systematic Reviews. 18. November 2015, doi:10.1002/14651858.CD010983.pub2 (wiley.com [abgerufen am 9. Juli 2020]).
- A. Engert u. a.: Involved-field radiotherapy is equally effective and less toxic compared with extended-field radiotherapy after four cycles of chemotherapy in patients with early-stage unfavorable Hodgkin’s lymphoma: results of the HD8 trial of the German Hodgkin’s Lymphoma Study Group. In: Journal of Clinical Oncology. Band 21, Nr. 19, 2003, S. 3601–3608. PMID 12913100
- V. Diehl u. a.: Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. In: NEJM. Band 353, Nr. 7, 2005, S. 744, PMID 12802024.
- R. Kuppers u. a.: Advances in biology, diagnostics, and treatment of Hodgkin’s disease. In: Biology of Blood and Marrow Transplantation. Band 12, Nr. 1, Supplement 1, Januar 2006, S. 66–76, PMID 16399588
- A. Josting u. a.: A new prognostic score based on treatment outcome of patients with relapsed Hodgkin lymphoma registered in the database of the German Hodgkin Lymphoma Study Group (GHSG). In: Journal of Clinical Oncology. Band 20, 2002, S. 221–230, PMID 11773173
- A. Josting u. a.: Prognostic factors and treatment outcome in primary progressive Hodgkin’s lymphoma – a report from the German Hodgkin’s Lymphoma Study Group (GHSG). In: Blood. Band 96, 2000, S. 1280–1286, PMID 10942369.
- Jahresbericht 2004 des Deutschen Kinderkrebsregisters, Universität Mainz (Memento vom 28. September 2007 im Internet Archive)
- Gerd Herold: Innere Medizin: eine vorlesungsorientierte Darstellung unter Berücksichtigung des Gegenstandskataloges für die Ärztliche Prüfung mit ICD 10-Schlüssel im Text und Stichwortverzeichnis. 2016. Auflage. Herold, Köln 2016, ISBN 978-3-9814660-5-8.
- Paul Lorigan, John Radford, Anthony Howell, Nick Thatcher: Lung cancer after treatment for Hodgkin’s lymphoma: a systematic review. In: The Lancet. Oncology. Band 6, Nr. 10, Oktober 2005, ISSN 1470-2045, S. 773–779, doi:10.1016/S1470-2045(05)70387-9, PMID 16198983.
- Lindsay M. Morton, Graça M. Dores, Rochelle E. Curtis, Charles F. Lynch, Marilyn Stovall: Stomach cancer risk after treatment for hodgkin lymphoma. In: Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. Band 31, Nr. 27, 20. September 2013, ISSN 1527-7755, S. 3369–3377, doi:10.1200/JCO.2013.50.6832, PMID 23980092, PMC 3770865 (freier Volltext).
- Dennis A. Eichenauer, Indra Thielen, Heinz Haverkamp, Jeremy Franklin, Karolin Behringer: Therapy-related acute myeloid leukemia and myelodysplastic syndromes in patients with Hodgkin lymphoma: a report from the German Hodgkin Study Group. In: Blood. Band 123, Nr. 11, 13. März 2014, ISSN 1528-0020, S. 1658–1664, doi:10.1182/blood-2013-07-512657, PMID 24478403.
- Dennis A. Eichenauer, Indra Thielen, Heinz Haverkamp, Jeremy Franklin, Karolin Behringer: Therapy-related acute myeloid leukemia and myelodysplastic syndromes in patients with Hodgkin lymphoma: a report from the German Hodgkin Study Group. In: Blood. Band 123, Nr. 11, 13. März 2014, ISSN 1528-0020, S. 1658–1664, doi:10.1182/blood-2013-07-512657, PMID 24478403.
- G. M. Dores u. a.: Second malignant neoplasms among long-term survivors of Hodgkin’s disease: a population-based evaluation over 25 years. In: Journal of Clinical Oncology. Band 20, Nr. 16, 2002, S. 3484–3494, PMID 12177110.
- Anthony J. Swerdlow, Craig D. Higgins, Paul Smith, David Cunningham, Barry W. Hancock: Second cancer risk after chemotherapy for Hodgkin’s lymphoma: a collaborative British cohort study. In: Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. Band 29, Nr. 31, 1. November 2011, ISSN 1527-7755, S. 4096–4104, doi:10.1200/JCO.2011.34.8268, PMID 21969511.
- Amit Sud, Hauke Thomsen, Kristina Sundquist, Richard S. Houlston, Kari Hemminki: Risk of Second Cancer in Hodgkin Lymphoma Survivors and Influence of Family History. In: Journal of Clinical Oncology. Band 35, Nr. 14, 10. Mai 2017, ISSN 0732-183X, S. 1584–1590, doi:10.1200/JCO.2016.70.9709, PMID 28384078, PMC 5455705 (freier Volltext).
- Andrea K. Ng, M. V. Patricia Bernardo, Edie Weller, Kendall Backstrand, Barbara Silver: Second malignancy after Hodgkin disease treated with radiation therapy with or without chemotherapy: long-term risks and risk factors. In: Blood. Band 100, Nr. 6, 15. September 2002, ISSN 0006-4971, S. 1989–1996, doi:10.1182/blood-2002-02-0634, PMID 12200357.
- Michael Schaapveld, Berthe M. P. Aleman, Anna M. van Eggermond, Cécile P. M. Janus, Augustinus D. G. Krol: Second Cancer Risk Up to 40 Years after Treatment for Hodgkin’s Lymphoma. In: New England Journal of Medicine. Band 373, Nr. 26, 24. Dezember 2015, ISSN 0028-4793, S. 2499–2511, doi:10.1056/NEJMoa1505949.
- F. E. van Leeuwen, W. J. Klokman, A. Hagenbeek, R. Noyon, A. W. van den Belt-Dusebout: Second cancer risk following Hodgkin's disease: a 20-year follow-up study. In: Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. Band 12, Nr. 2, Februar 1994, ISSN 0732-183X, S. 312–325, doi:10.1200/JCO.1994.12.2.312, PMID 8113838.
- T. Hodgkin: On some morbid appearances of the absorbent glands and spleen. In: Medico-Chirurgical Transactions. (London) Band 17, 1832, S. 68–114, PMID 4630498.
- C. Sternberg: Über eine eigenartige unter dem Bilde der Pseudoleukämie verlaufende Tuberculose des lymphatischen Apparates. In: Zeitschrift für Heilkunde. Band 19, 1898, S. 21.
- D. M. Reed: On the pathological changes in Hodgkin’s disease, with special reference to its relation to tuberculosis. In: Johns Hopkins Hosp Rep. Band 10, 1902, S. 133.
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 376–449, hier: S. 434–437: Die Lymphogranulomatose (malignes Granulom, Sternbergsche Krankheit, Hodgkinsche Krankheit).
- P. Calabresi, B. A. Chabner: Antineoplastic Agents. In: A. G. Gilman, T. W. Rall, A. S. Nies, P. Taylor (Hrsg.): Goodman and Gilman’s the pharmacological basis of therapeutics. 8th international edition. McGraw-Hill Health Professions Division. New York/ St. Louis/ San Francisco 1992, ISBN 0-07-112621-X.
- C. P. J. Adair, H. J. Bagg: Experimental and clinical studies on the treatment of cancer by dichloroethylsulphide (mustard gas). In: Annals of Surgery. Band 93, 1931, S. 190.
- Janusz Piekałkiewicz: Die Schlacht von Monte Cassino. Zwanzig Völker ringen um einen Berg. Bechtermünz Verlag, Augsburg, ISBN 3-86047-909-1, S. 66/67.
- L. S. Goodman u. a.: Use of methyl-bis(beta-chloroethyl)amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia. In: JAMA. Band 132, 1946, S. 126, PMID 6368885
- V. T. DeVita Jr. u. a.: Combination chemotherapy in the treatment of advanced Hodgkin’s disease. In: Annals of Internal Medicine. Band 73, 1970, S. 881, PMID 5525541
- M. Medetti: Nitrogen mustards in the treatment of children. In: Osp Maggiore. Band 40, Nr. 1, Januar 1952, S. 38–40, PMID 14941617.