Harlekin-Ichthyose
Die Harlekin-Ichthyose ist eine sehr seltene angeborene Hauterkrankung und stellt die schwerste Form der autosomal-rezessiven kongenitalen Ichthyosen (ARCI) dar. Die gestörte Hornschichtbildung der Oberhaut zeigt sich hierbei oft schon vorgeburtlich mit dem Hauptmerkmal einer panzerartigen Bedeckung des ganzen Körpers durch Hornschuppen.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q80.4 | Ichthyosis congenita gravis [Harlekinfetus] |
| ICD-10 online (WHO-Version 2019) | |
Die Erstbeschreibung stammt aus dem Jahre 1880 von Robert William Smith (1807–1873).[3][4] Synonyme sind: Ichthyosis congenita gravis; Harlekinfetus; Ichthyosis congenita Riecke I; Keratosis diffusa maligna; Ichthyosis congenita fetalis; Hyperkeratosis universalis congenita; Ichthyosis congenita universalis; Harlequin ichthyosis; Ichthyose, kongenitale, Typ Harlequin; Ichthyosis fetalis vom Harlekin Typ.
Die Erkrankung ist nicht zu verwechseln mit dem Harlequin-Syndrom.
Verbreitung
Die Häufigkeit wird mit unter 1 zu 1.000.000 angegeben, die Vererbung folgt einem autosomal-rezessiven Erbgang.[1]
Ursache
Der Erkrankung liegen Mutationen im ABCA12-Gen auf Chromosom 2 (Genort q35) zugrunde, das für einen ABC-Transporter kodiert.[5]
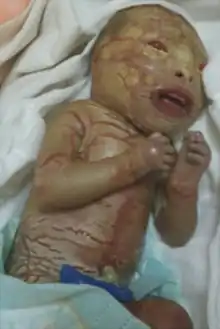
Klinische Erscheinungen
Wichtige Kriterien für die Diagnose anhand des klinischen Bildes sind:[1][2]
- bei Geburt in eine kollodiumähnlich steife schimmernde Membran eingeschlossen, die wie eine zusätzliche Hautschicht erscheint – sogenannte Kollodium-Membran
- große, dicke, plattenartige Schuppen auf dem gesamten Körper, dazwischen tiefe Hauteinrisse
- starke Ausstülpung der Augenlider (Ektropium) und der Lippen (Eklabium) – sogenannter Harlekin-Aspekt
- eingeschränkte Beweglichkeit der Extremitäten und des Brustraumes
- Mund- und Genitalschleimhaut befallen
- Kontrakturen
Diagnose
Die Diagnose ergibt sich aus dem klinischen Bild. Die vorgeburtliche Sonografie kann einen Verdacht nahelegen, der durch eine humangenetische Untersuchung gesichert wird.[6]
Differentialdiagnose
Abzugrenzen sind:[1]
Heilungsaussicht
Viele der Betroffenen sterben kurz nach der Geburt infolge einer Ateminsuffizienz oder einer Sepsis. Im späteren Verlauf ist die Hautpanzerung leicht rückläufig, es kommt zu Schuppenbildung und Erythrodermie.[2]
Fortschritte der Intensivmedizin haben die Letalität gesenkt.[7]
Literatur
- K. Washio, M. Sumi, K. Nakata, A. Fukunaga, K. Yamana, T. Koda, I. Morioka, C. Nishigori, K. Yamanishi: Case of harlequin ichthyosis with a favorable outcome: Early treatment and novel, differentially expressed, alternatively spliced transcripts of the ATP-binding cassette subfamily A member 12 gene. In: The Journal of dermatology. Bd. 44, Nr. 8, August 2017, S. 950–953, doi:10.1111/1346-8138.13823, PMID 28295493.
- H. Ahmed, E. A. O'Toole: Recent advances in the genetics and management of harlequin ichthyosis. In: Pediatric Dermatology. Bd. 31, Nr. 5, 2014 Sep-Oct, S. 539–546, doi:10.1111/pde.12383, PMID 24920541 (Review).
Einzelnachweise
- Harlekin-Ichthyose. In: Orphanet (Datenbank für seltene Krankheiten).
- Enzyklopädie Dermatologie
- Who named it
- R. W. Smith: A case of intrauterine ichthyosis. In: American Journal of Obstetrics and Gynecology, 1880, Bd. 13, S. 458–461.
- Ichthyosis, congenital, autosomal recessive 4B (harlequin). In: Online Mendelian Inheritance in Man. (englisch)
- S. Rathore, L. S. David, M. M. Beck, M. S. Bindra, G. Arunachal: Harlequin Ichthyosis: Prenatal Diagnosis of a Rare Yet Severe Genetic Dermatosis. In: Journal of clinical and diagnostic research : JCDR. Bd. 9, Nr. 11, November 2015, S. QD04–QD06, doi:10.7860/JCDR/2015/15250.6705, PMID 26675324, PMC 4668483 (freier Volltext).
- J. B. Glick, B. G. Craiglow, K. A. Choate, H. Kato, R. E. Fleming, E. Siegfried, S. A. Glick: Improved Management of Harlequin Ichthyosis With Advances in Neonatal Intensive Care. In: Pediatrics. Bd. 139, Nr. 1, Januar 2017, S. , doi:10.1542/peds.2016-1003, PMID 27999114 (Review).