β-Glucuronidase
β-Glucuronidase (GUSB) ist der Name für Enzyme, die β-Glucuronide spalten, also Glykoside, die sich von Glucuronsäure ableiten. Dieser Reaktionsschritt ist unverzichtbar für Tiere und manche Bakterien beim Abbau der Substanzen Häm, Chondroitinsulfat und Keratansulfat. Das Enzym findet sich in den Lysosomen von Leber, Milz und Mandeln. Defekte im GUSB-Gen beim Menschen können Mucopolysaccharidose Typ 7 verursachen und spielen eine Rolle bei Hydrops fetalis.[1][2]
| β-Glucuronidase | ||
|---|---|---|
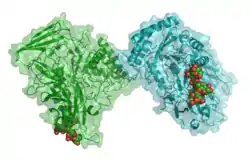
| ||
| Bänder-/Oberflächenmodell des Dimers mit Nonaglykosid nach PDB 1BHG | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 629 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Homotetramer | |
| Isoformen | Long, Short | |
| Bezeichner | ||
| Gen-Name | GUSB | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 3.2.1.31, Glykosidase | |
| Reaktionsart | Hydrolyse | |
| Substrat | β-D-Glucuronosid + H2O | |
| Produkte | D-Glucuronat + Alkohol | |
| Vorkommen | ||
| Homologie-Familie | Beta-Glucuronidase | |
| Übergeordnetes Taxon | Lebewesen | |
| Ausnahmen | Pflanzen | |
Pathologie
Im Falle eines Funktionsmangels an β-Glucuronidase kommt es zu einer Erbkrankheit, einem Beta-Glucuronidasemangel, der als Sly-Syndrom bezeichnet wird und zu den Mukopolysaccharidosen gehört (Typ VII).[3]
Verwendung
Sie findet Anwendungen in der Biotechnologie als Reportergen. Ihre Aktivität lässt sich mit verschiedenen Methoden nachweisen:
- β-Glucuronidase hydrolysiert X-Gluc, wodurch ein blauer Farbstoff entsteht;
- 4-Methylumbelliferyl-β-D-glucuronid (MUG) wird zu 4-Methylumbelliferon (MU) hydrolysiert, welches durch seine Fluoreszenz nachweisbar ist. Dabei kann über die Intensität dieser Fluoreszenz auf die Umsatzrate der β-Glucuronidase geschlossen werden.
- β-Glucuronidase spaltet p-Nitrophenyl-β-D-glucuronid in Glucuronsäure und p-Nitrophenol. p-Nitrophenol dissoziiert durch eine Base in p-Nitrophenolat, welches gelb gefärbt ist.
Siehe auch
Einzelnachweise
- UniProt-Eintrag
- Eintrag zu β-Glucuronidase. In: Römpp Online. Georg Thieme Verlag, abgerufen am 29. Dezember 2014.
- Mukopolysaccharidose Typ 7. In: Orphanet (Datenbank für seltene Krankheiten).