Ghost lineage
Als ghost lineage (deutsch etwa „Geisterhafte Abstammung“) werden in der Paläontologie und Phylogenetik Abstammungslinien bezeichnet, deren Vorhandensein in einer bestimmten Zeitepoche (oder stratigraphischen Einheit) nicht direkt nachgewiesen, sondern nur indirekt erschlossen worden ist. Das Konzept und der Ausdruck gehen auf den Paläontologen Mark Norell zurück.[1] Für die Dauer einer bestimmten Linie wird analog auch von „ghost range“ gesprochen. Ghost lineages sind grundsätzlich problematisch, weil sie jeweils einer zusätzlichen Hilfshypothese entsprechen. Ihre Heranziehung in der Forschung ist aber in einigen Bereichen unvermeidlich. In der Praxis wird – gemäß Ockhams Rasiermesser – bei der Interpretation der Daten in der Regel die Hypothese bevorzugt, die weniger oder zumindest kürzere ghost lineages zur Folge hat.[2]


Ghost lineages entstehen infolge einer Reihe unterschiedlicher Methoden und theoretischer Ableitungen. Meist beruhen sie darauf, dass auf dasselbe Problem, z. B. das Alter oder die Verwandtschaftsverhältnisse einer bestimmten Gruppe von Organismen, unterschiedliche und voneinander unabhängige Methoden angewandt werden. Unterschiedliche Altersschätzungen beruhen vor allem auf absolut datierten Fossilien, mit den Methoden der molekularen Uhr abgeleitete Altersschätzungen oder aus der kladistischen Analyse der Verwandtschaftsverhältnisse einer Gruppe erschlossene Reihenfolgen von Aufspaltungsereignissen evolutionärer Stammlinien.
Interpolationen
Der einfachste Fall der Erzeugung von ghost lineages beruht direkt auf der Unvollständigkeit der fossilen Überlieferung. Wird ein bestimmtes Taxon (eine Art, eine Gattung oder eine höhere taxonomische Einheit) in einer Lagerstätte einer geologischen Zeitepoche und in einer anderen gefunden, die ein davon abweichendes Alter aufweist, muss das Taxon in der dazwischenliegenden Zeit ebenfalls existiert haben. In der Paläontologie wird die Anwendung dieses naheliegenden Prinzips oft als „range through method“ bezeichnet.[3] Genauso kann ein Taxon sowohl fossil als auch lebend (rezent oder auch extant) nachgewiesen sein. Ein prominentes Beispiel hierfür sind die Quastenflosser, die nur bis in die Kreidezeit fossil überliefert sind und bis zu den heute lebenden Vertretern eine Lücke von über 70 Millionen Jahren aufweisen.
Phylogenetische Verzweigungen
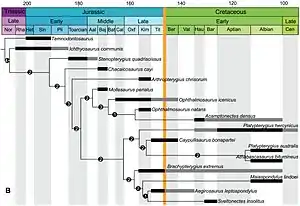
Während auf Interpolation beruhende ghost lineages die Existenzdauer eines Taxons nur quasi auffüllen, kann sie durch die Verwendung phylogenetischer Verzweigungsmuster (oder Kladogramme) auch über den fossil dokumentierten Bereich hinaus verlängert werden. Im Extremfall kann so das Alter von Gruppen abgeschätzt werden, von denen gar keine Fossile gefunden wurden.
Die Methode beruht auf der Anwendung von Kladogrammen im Rahmen der phylogenetischen Systematik oder Kladistik. Hierbei wird anhand von ursprünglichen Merkmalen, die verschiedene Mitglieder der Gruppe aufgrund gemeinsamer Vererbung von einer Stammart gemeinsam haben (den sogenannten Autapomorphien) die Topologie und Reihenfolge der Kladogenese erschlossen. Dies kann, obwohl auch Fossilien verwendet werden können, auch ausschließlich mit lebenden Arten durchgeführt werden.
Da alle lebenden Arten eine gemeinsame Abstammung besitzen, können neue Taxa ausschließlich dadurch entstehen, dass bestehende ihre Form und Merkmale verändern und sich schließlich in mehrere neue Taxa aufspalten. Zwei miteinander verwandte Linien, die durch die Aufspaltung einer solchen Stammlinie neu entstanden sind, müssen notwendigerweise untereinander gleich alt sein. Ist nun etwa durch datierte Fossilien das Alter einer dieser Linien bekannt, muss die andere Linie mindestens ebenso alt sein.[4] Verlängerung mit dieser Methode ist naturgemäß nicht über das älteste Fossil der ältesten Linie hinaus möglich.
Molekulare Uhren
Neben der Datierung von Fossilien kann das Alter einer Stammlinie unabhängig davon durch die Methode der molekularen Uhr bestimmt werden. Dabei wird über den Unterschied zweier DNA-Sequenzen und einer mittleren Mutationsrate das Alter der Verzweigung abgeschätzt. Nach der neutralen Theorie der molekularen Evolution sollte die Sequenzveränderung in etwa proportional zur verstrichenen Zeit sein, so dass bei Kenntnis der Veränderungsrate das Alter direkt ablesbar wäre.
In der Praxis gestaltet sich diese Methode oft schwierig, weil die Mutationsrate je nach Gruppe und Sequenz oft deutlich variiert und das Alter der Kladen dadurch falsch eingeschätzt wird. Die Kalibrierung der jeweiligen Molekularen Uhren über die Referenz von Fossilien oder Tektonikereignissen ist daher aufwendig und hat starken Einfluss auf die Länge so errechneter ghost lineages.
Abschätzung früherer Biodiversität
Ghost lineages müssen auch berücksichtigt werden, wenn man die Biodiversität der Flora und Fauna früherer Epochen ermitteln will.[5][6] Diese ist nur für eine Handvoll von Lagerstätten so gut fossil dokumentiert, dass sie mit einiger Verlässlichkeit direkt aus dem Fossilbericht abgelesen werden könnte. Will man herausfinden, ob in einer bestimmten Epoche z. B. die Artenzahl zu- oder abnahm, ob ein bestimmtes geologisches Ereignis im Zusammenhang mit einer adaptiven Radiation gestanden haben könnte oder ob ein vermeintliches Massenaussterben real war oder nur durch Zufälle der Überlieferung vorgetäuscht, ist es erforderlich, den Artbestand der entsprechenden Epoche zu rekonstruieren. Hierzu müssen die ghost lineages einbezogen werden, da ansonsten die Artenzahl massiv unterschätzt würde.
Probleme
Da ghost lineages definitionsgemäß ausschließlich indirekt ermittelte Konstrukte sind, beruhen alle Hypothesen, die auf ihre Existenz gegründet werden, in kritischer Weise darauf, dass die der Konstruktion zugrunde liegenden Hypothesen korrekt sind. Wird ein Fossil falsch datiert, ein Kladogramm unzutreffend konstruiert, eine molekulare Uhr falsch kalibriert, werden dadurch umfangreiche ghost lineages erzeugt, deren (schattenhafte) Existenz mit der Anwendung korrekter Methoden in sich zusammenfällt. Fossilien können falsch datiert oder, häufiger, schlecht erhaltene oder unvollständige Fossilien irrtümlich oder aus Wunschdenken einer bekannten Linie zugeordnet werden.
Das Alter korrekt datierter und zugeordneter Fossilien muss zwangsläufig immer das Alter der Stammlinie unterschätzen (da ja nicht ausgerechnet deren erster Vertreter fossiliert worden sein wird). Andererseits gibt es ernstzunehmende Hinweise darauf, dass die Methode der molekularen Uhr das Alter von Stammlinien in aller Regel überschätzt. Außerdem beruht diese Methode auf Kalibrierung von kritischen Verzweigungsereignissen, die sowohl fossil falsch datiert wie auch phylogenetisch falsch gedeutet sein könnten (vgl.[7][8][9]).
Quellen
Literatur
- Demetrio Boltovskoy: The Range-through Method and First-Last Appearance Data in Paleontological Surveys. In: Journal of Paleontology 62 (1), 1998. S. 157–159.
- Lionel Cavin, Peter L. Forey: Using Ghost Lineages to Identify Diversification Events in the Fossil Record. In: Biology Letters 3 (2), 2007. doi:10.1098/rsbl.2006.0602, S. 201–204.
- Mark A. Norell: Tree-based Approaches to Understanding History: Comments on Ranks, Rules and the Quality of the Fossil Record. In: American Journal of Science 293-A, 1993. S. 407–417.
- Alexandr P. Rasnitsyn: Testing Cladograms by Fossil Record: The Ghost Range Test. In: Contributions to Zoology 69 (4), 2000. S. 251–258, doi:10.1163/18759866-06904003.
Weblinks
- Matt Wedel: Ghost lineages. University of California Museum of Paleontology, www.ucmp.berkeley.edu, Mai 2010.
Einzelnachweise
- M.A. Norell (1992): Taxic origin and temporal diversity: the effect of phylogeny. In: M. J. Novacek, Q. D. Wheeler (eds.): Extinction and phylogeny. Columbia University Press, New York: 88–118.
- Rasnitsyn 2000, S. 251.
- Boltovskoy 1998, S. 157–159.
- Norell 1993, S. 409.
- Cavin & Forey 2007, S. 201.
- Andrew B. Smith (2003): Getting the measure of diversity. Paleobiology, 29(1): 34–36.
- Philip C.J. Donoghue, Michael J. Benton (2007): Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends in Ecology and Evolution, Vol. 22, No. 8
- Gregory A Wray (2001): Dating branches on the Tree of Life using DNA. Genome Biology 2001, 3
- Michael J. Benton, Francisco J. Ayala (2003): Dating the Tree of Life. Science 300: 1698-1700.