Morbus Gaucher
Morbus Gaucher [], auch Gaucher-Syndrom und Gauchersche Krankheit, ist eine seltene Erbkrankheit und die häufigste der lysosomalen Speicherkrankheiten, einer Störung des Lipidstoffwechsels. Bei der autosomal-rezessiv vererbten Krankheit liegt eine Mutation im Gen der β-Glucocerebrosidase (EC 3.2.1.45; Chromosom 1; Genlocus 1q21; Gensymbol GBA) vor; bekannt sind über 400 Varianten[1]. Es kommt dadurch zu einer verringerten Aktivität dieses in den Lysosomen lokalisierten Enzyms, so dass Glucocerebrosid (zuckerhaltige Fettstoffe) nur noch unzureichend in Glukose und Ceramide aufgespalten werden. Glukocerebroside sind z. T. selbst direkter Bestandteil von Zellmembranen (insbesondere der Erythrozyten) vor allem aber Zwischenprodukte beim Auf- und Abbau zahlreicher weiterer, komplexerer Glykolipide der Zellmembranen. Durch den gestörten Abbau reichern sie sich daher insbesondere in den Fresszellen des Körpers (Makrophagen) und in Monozyten an, die in der Folge zu sogenannten Gaucherzellen werden. Diese setzen unter anderem zahlreiche Zytokine und Faktoren frei die letztlich die Krankheitssymptome bestimmen: Eine (zum Teil massive) Vergrößerung der Milz (Splenomegalie), eine Vergrößerung der Leber (Hepatomegalie), Störung der Blutbildung im Knochenmark mit einem Mangel an Blutplättchen (Thrombozytopenie), roten Blutkörperchen (Anämie) und weißen Blutkörperchen (Leukozytopenie). Der Befall des Knochenmarks und des gesamten Skelettsystems (insbesondere der Hüfte und des Oberschenkels) führt zur Zerstörung des Knochens, die sich mitunter in Form fieberhafter schmerzhafter Schübe, sogenannter Knochenkrisen, manifestiert. Bisweilen sind auch weitere Organsysteme wie das Nervensystem oder die Lunge betroffen.
| Klassifikation nach ICD-10 | |
|---|---|
| E75.2 | Sonstige Sphingolipidosen Gaucher-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Oft findet sich eine durch den körperlichen Befund allein nicht erklärbare Erschöpfbarkeit der Betroffenen, so dass diese zunächst als Simulanten oder Hypochonder verkannt werden. Patienten mit Morbus Gaucher haben ein deutlich erhöhtes Risiko in der Folge ein Malignom (vor allem des Blutes bzw. lymphatischen Systems wie Multiples Myelom, Non-Hodgkin-Lymphome und der Leber (Leberzellkarzinom)) oder einen Morbus Parkinson zu entwickeln. Da die Erkrankung selten ist und das Spektrum der Symptome mit verschiedenen anderen, häufigeren Erkrankungen überlappt, dauert es auch heute noch oft viele Jahre, bis die korrekte Diagnose gestellt wird. Mit der Enzymersatztherapie (eine Infusionstherapie) und der Substratreduktionstherapie (orale Therapie mit Kapseln) sind zwei wirksame Behandlungsoptionen verfügbar.
Benannt ist die Erkrankung (lat.: Morbus) nach Philippe Gaucher (1854–1918), einem französischen Dermatologen, der die vermehrten Speicherzellen in der Milz 1882 erstmals beschrieb. Seit 1934 ist bekannt, dass sie Glucocerebroside enthalten.
Einteilung
In der Vergangenheit wurde der M. Gaucher sehr streng in drei Typen eingeteilt: Morbus Gaucher Typ 1, Typ 2 und Typ 3. Diese Einteilung basierte auf dem Zeitpunkt des Krankheitseintritts, den jeweiligen Symptomen, der Mitbeteiligung des Nervensystems und der Lebenserwartung der Patienten. Diese Trennung wird zunehmend verlassen, unter anderem da es Übergangsformen gibt, die nicht eindeutig einem Typ zuzuordnen sind. Heute unterscheidet man zunehmend in eine neuronopathische und eine nicht-neuronopathische Verlaufsform, d. h. das Auftreten bzw. Fehlen von Nervenschädigungen entscheidet über die Zuordnung zu den beiden Hauptgruppen. Diese historische Klassifikation ist aber weiterhin sehr gebräuchlich.
Morbus Gaucher Typ I (auch nicht neuronopathischer M. Gaucher); ca. 94 % der Patienten
Dies ist die häufigste Form von Morbus Gaucher. Die Erstmanifestation kann in jedem Alter auftreten. Bei dieser Form ist das Nervensystem nicht involviert (nicht neuronopathisch). Der Verlauf der Erkrankung kann sehr unterschiedlich sein, einige Patienten haben kaum Symptome und führen ein weitgehend normales Leben, während andere unter vielfältigen und schweren Symptomen leiden. Die Genotyp-Phänotyp-Korrelation ist dabei nur sehr gering.[2]
Morbus Gaucher Typ II (auch akut-neuronopathischer M. Gaucher); ca. 1 % der Patienten
Dieser Typ ist sehr selten und tritt nur im Kindesalter auf. Aufgrund der schweren neurologischen Beteiligung werden betroffene Kinder selten älter als 2 Jahre. Für diesen sind die verfügbaren therapeutischen Optionen ohne relevanten Einfluss auf den Verlauf. Manche Autoren grenzen hiervon noch eine perinatal letale Verlaufsform ab.[2]
Morbus Gaucher Typ III (auch chronisch-neuronopathische Verlaufsform); ca. 5 % der Patienten
Dieser Typ ist ebenfalls selten und manifestiert sich meist in der frühen bis späten Kindheit, die Symptome sind mittel bis schwer, der Krankheitsverlauf ist progredient. Bei diesen Patienten kann die Lebenserwartung vermindert sein.[3]
Unterteilung in Subtypen
Manche Autoren unterscheiden hierbei noch Subtypen:[3]
- Morbus Gaucher Typ IIIA: Charakterisiert durch Myoklonien und Demenz
- Morbus Gaucher Typ IIIB: Charakterisiert durch frühen Beginn einer isolierten supranukleären, horizontalen Blicklähmung und einen aggressiven Verlauf
- Morbus Gaucher Typ IIIC: Charakterisiert durch kardiovaskuläre Verkalkungen
Häufigkeit
Die betreffende Gendefekte werden mit Ausnahme einiger Fälle des Typs 2 autosomal-rezessiv vererbt. Die Erkrankung tritt in regional sehr unterschiedlicher Häufigkeit auf. In Westeuropa ist vermutlich 1 von 40.000–60.000 Menschen betroffen. Dagegen sind in der aschkenasisch-jüdischen Bevölkerung mit einer von 1.000 Personen relativ betrachtet deutlich mehr Menschen betroffen. Die Häufigkeit der heterozygoten Mutation wird dabei auf 1:30 geschätzt. Damit ist der Morbus Gaucher die häufigste lysosomale Speicherkrankheit. Auch innerhalb der türkischen Bevölkerung ergibt sich ein gegenüber Westeuropäern erhöhtes Auftreten dieser Mutationen.[4][5]
Ursache
Der menschliche Körper benötigt das Enzym Glucocerebrosidase (im deutschen Sprachraum auch: Glukozerebrosidase), um gealterte Zellmembranbestandteile abzubauen. Ein Mangel wird daher besonders auffällig bei Zellen mit kurzer Lebenszeit, so den weißen und roten Blutzellen. Ohne ausreichende Aktivität des Enzyms lagern sich die Membranbestandteile in den Lysosomen insbesondere der Makrophagen ab. Je nach Art der Mutation des für das Enzym kodierenden Gens kommt es zu mehr oder weniger starkem bis vollständigem Funktionsausfall des Enzyms.[4] Mittlerweile sind mehr als 400 verschiedene Mutationen beschrieben.[6][7]
Symptome
Die Schwere des Enzymdefekts bestimmt das Alter bei ersten Symptomen und die Organe, an denen die Symptome vor allem auftreten:
Beim nicht-neuronopathischen Typ (früher Typ I) ist die Enzymaktivität noch relativ hoch. Erste Symptome können auch erst im Erwachsenenalter auftreten, der Verlauf kann milde sein. Es kommt zu Veränderungen vor allem an inneren Organen („viszeraler Typ“) in Form einer extremen Vergrößerung der Leber und Milz. Durch die Milzvergrößerung resultiert ein gesteigerter Abbau von Blutzellen. Dadurch kommt es zu einer Blutarmut und einem Blutplättchenmangel. Daraus folgt ein erhöhtes Blutungsrisiko und eventuell Kreislaufprobleme. Häufig ist auch das Skelett beteiligt. Dies äußert sich in chronischen aber plötzlich auftretenden, schweren Schmerzen, insbesondere an den Hüftgelenken, die von Fieber und Entzündungszeichen begleitet sein können. Im Rahmen der Knochenschädigung kann es auch zu Osteolysen kommen.[8]
Beim akut-neuronopathischen Typ (früher Typ II) ist die Enzymaktivität besonders gering, schon Säuglinge zeigen schwere Störungen des Nervensystems mit geistiger Behinderung, Krämpfen und ausgeprägten Gedeihstörungen. Die Kinder sterben meist vor Ende des zweiten Lebensjahres. Typ II kann zudem auch im dominanten Erbgang auftreten.[4]
Beim chronisch-neuronopathischen Typ (früher Typ III) liegen die Enzymaktivitäten meist zwischen Typ I und Typ II. Es kommt etwa ab dem zweiten Lebensjahr zu Nervenschädigungen und Gedeihstörungen.[5] Diese Form tritt gehäuft in Schweden auf.[6]
Diagnose
Leitsymptome ist die Vergrößerung der Milz (meist mittels Ultraschalluntersuchung), die sich im Verlauf bei praktisch allen Patienten findet, oft gepaart mit einer Vergrößerung der Leber. Häufig finden sich mehr oder minder ausgeprägte Veränderungen des Blutbildes, insbesondere ist die Zahl der Thrombozyten oft verringert. Der Befall der Knochen wird vor allem in der Kernspintomographie deutlich, wohingegen ein konventionelles Röntgenbild oft noch keine Veränderungen zeigt. Allerdings können sich auch hier typische Manifestationen wie die sog. Erlenmeyerdeformität finden.
Im Routinelabor findet sich oft eine Erhöhung des Ferritin, des ACE und der sauren nicht-tartrathemmbare Phosphatase. Dies ist jedoch sehr unspezifisch, so dass bei Verdacht oder zum Ausschluss ein gezielter Enzymtest zum Nachweis einer verringerten β-Glukozerebrosidase-Aktivität in Leukozyten (aus EDTA-Vollblut oder mittels Trockenbluttest (DBS)) – eventuell ergänzt durch die Bestimmung von Glucosylsphingosin (lyso-Gb1/lysoGL1) – erfolgen sollte. Die bisweilen durchgeführte Knochenmarkpunktion kann bei bis zu einem Drittel der Fälle falsch negativ sein. Sie ist daher zum Nachweis oder Ausschluss eines Morbus Gaucher heute obsolet.
Liegt ein Verdacht auf Morbus Gaucher vor, wird im Labor die Enzymaktivität der beta-Glukozerebrosidase im Blut bestimmt. Ist sie erniedrigt, so ist die Diagnose Morbus Gaucher gesichert. Zusätzlich wird heute fast immer auch eine genetische Analyse durchgeführt.
Sowohl für die Messung der Enzymaktivität als auch für die genetische Analyse steht heute ein einfach in den Praxisalltag zu integrierender Trockenbluttest (Dried Blood Spot, DBS) zur Verfügung: Dafür werden einige Tropfen Blut auf eine Trockenblutkarte aufgetropft. Nachdem sie getrocknet sind, wird die Karte per Post an ein spezialisiertes Labor geschickt. Dort wird das Blut wieder aus der Filterkarte herausgelöst und für die folgenden Tests aufbereitet.
Zur Bestimmung der Enzymaktivität wird zu einer definierten Menge Blut eine definierte Menge Substrat dazugegeben. Nach einer bestimmten Zeit wird z. B. per Massenspektrometrie analysiert, wie viel Produkt durch die Enzymreaktion entstanden ist. Hieraus lässt sich schließen, wie aktiv das Enzym ist. Um die Verlässlichkeit der Messwerte zu gewährleisten, ist es wichtig, dass ein zertifizierter Assay verwendet wird.
Für die genetische Analyse wird das Gen der beta-Glukozerebrosidase sequenziert. Beide Tests – die Messung der Enzymaktivität und die genetische Analyse – kann – je nach Labor - aus dem Material einer Trockenblutkarte erfolgen.[9]
Behandlung
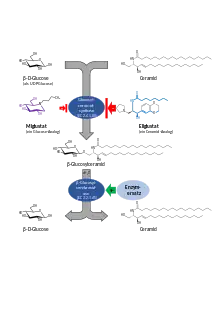
Durch den Mangel an Glucocerebrosidaseaktivität bei Morbus Gaucher kommt es zu einem Ungleichgewicht zwischen Auf- und Abbau von Glucosylceramid (oft als GL1 oder Gb1 abgekürzt). Insbesondere beim Typ 1, der nicht neuronopathischen Verlaufsform, und zum Teil beim Typ 3, kann die Einlagerung von GL1 und damit die Erkrankung mittels zweier unterschiedlicher Therapieprinzipien – Enzymersatztherapie und Substratreduktionstherapie – sehr gut behandelt werden. Daher wird die früher bisweilen eingesetzte Stammzelltransplantation – durch die Makrophagen ohne Enzymdefekt gebildet werden – aufgrund der damit verbundenen erheblichen Risiken kaum mehr eingesetzt.
Enzymersatztherapie (EET oder ERT)
Der ursächliche Mangel an Glucocerebrosidaseaktivität (EC 3.2.1.45) lässt sich durch lebenslange, in der Regel zweiwöchentliche Infusion biotechnologisch hergestellter Glucocerebrosidase ausgleichen. In Europa wurden Imiglucerase und Velaglucerase alfa zugelassen und verfügbar. Aufgrund der bereits 1994 in den USA erfolgten Zulassung lagen 2018 für Imiglucerase die meisten Daten und längsten praktischen Erfahrungen vor (Erstzulassung Velaglucerase alfa: 2010[10]; In Europa nicht zugelassene Taliglucerase alfa: 2012[11]).
Die Aufnahme erfolgt über terminale Mannose-Reste Glykosylierungen, wodurch das infundierte Enzym bevorzugt in (beim Morbus Gaucher auch hauptsächlich betroffenen) Makrophagen aufgenommen wird, so deren Lysosomen erreicht und dort die Speichersubstanz Glucosylceramid abbauen kann.[8]
Substratreduktionstherapie (SRT)
Das der Speicherung von Glucosylceramid zugrunde liegende Ungleichgewicht zwischen Auf- und Abbau lässt sich auch durch eine (partielle) Hemmung der Glucosylceramidsynthese bzw. des dafür verantwortlichen Enzymes Glucosylceramidsynthase kompensieren. Es wird weniger Glucosylceramid gebildet, es liegt also weniger Substrat für das bei M. Gaucher unzureichend vorhandene oder aktive Enzym Glucocerebrosidase vor, das diese abbauen müsste. Entsprechende Inhibitoren sind zumeist kleine Moleküle und lassen sich auch als oral als Kapsel verabreichen, wodurch dem Patienten die zeitaufwendigen Injektionen und die erforderliche Terminplanung erspart bleibt.
Das erste zur Substratreduktiontherapie für Morbus Gaucher zugelassene Präparat war Miglustat (Erstzulassung: 2002). Miglustat ist ein Iminozucker und ein Analogon der Glucose. Es bindet an dessen Bindungsstelle und verhindert so die Synthese von Glucosylceramid. Da Glucose an vielen Vorgängen im Körper beteiligt ist, werden auch weitere Enzyme durch Miglustat gehemmt. Dazu zählen unter anderm auch einige Zuckerspaltende Enzyme im Darm, was in der Folge zu belastenden Nebenwirkungen wie (osmotisch bedingten) Durchfall führen kann, aber auch zentralnervöse Symptome. Die Anwendung von Miglustat ist daher auf Patienten mit einem leichten bis mittelschweren, nicht-neuronopathischen Morbus Gaucher, für die die Enzymsubstitutionstherapie ungeeignet ist, beschränkt (Zweitlinientherapie).
Das zweite, im Januar 2015 zugelassene, Präparat zur Substratreduktionstherapie Eliglustat ist ein Analogon des Ceramids. Es ist wesentlich spezifischer und selektiver für die Glucosylceramidsynthase als Miglustat, so dass es wesentlich besser verträglich ist. Beispielsweise waren Magen-Darm-Beschwerden in den Zulassungsstudien in der Placebogruppe deutlich häufiger. Anders als Miglustat gelangt es kaum in ZNS, so dass auch hier kaum Nebenwirkungen beobachtet wurden. Die Wirksamkeit ist sowohl bei bisher unbehandelten[12], wie auch mit Enzymersatztherapie vorbehandelten Patienten einer Enzymersatztherapie vergleichbar.[13] Langzeitdaten über 8 Jahre belegen zudem, dass insbesondere schwer betroffene Gaucherpatienten in besonderem Maße profitieren[14]. Eliglustat wird über das Cytochrom-P450-System, insbesondere CYP2D6 und zu einem kleineren Teil auch über CYP3A4, abgebaut. Die Aktivität dieser Enzymsysteme ist individuell verschieden, so dass vor der Verordnung eine einmalige genetische Bestimmung des entsprechenden Stoffwechseltyps erfolgen muss. Eliglustat ist für erwachsene Patienten mit Typ 1 zugelassen, die bezogen auf CYP2D6 langsame, intermediäre oder schnelle Metabolisiere sind[15].
Zur Therapiekontrolle eignen sich neben klinischen Parametern, Kernspintomographie, Ultraschall und Blutbild auch die Aktivität bzw. Konzentrationen von Chitotriosidase, Glucosylsphingosin (= lyso-Gb1 oder lyso-GL1) bzw. CCL18.[16]
Therapiekontrolle
Die Therapiekontrolle erfolgt – neben den klinischen Parametern – mittels Bildgebung (Kernspintomographie, Ultraschall, ggf. Röntgen und Knochendichtemessung) und Laboruntersuchungen. Neben dem Blutbild gilt die Aktivität des Enzyms Chitotriosidase als Marker für die Speicherlast. Die Messung ist jedoch nicht wenig standardisiert und laborspezifisch. Zudem ist eine Erhöhung nicht Gaucher-spezifisch und etwa 5–10 % der Patienten verfügen über keine Chitotriosidaseaktivität. Daher etabliert sich zunehmend Glucosylsphingosin (= lyso-Gb1 oder lyso-GL1) als neuer Monitoringparameter.
Aussichten
Die Prognose hängt von Typ und Schwere der Erkrankung, von der Verfügbarkeit und vom tatsächlichen Einsatz der Behandlung ab: Endgültige Heilung kann nur eine Beseitigung des Gendefekts durch Gentherapie bringen. Bisher ist eine solche Lösung allerdings noch nicht in Sicht. Bis dahin ist das Ziel eine möglichst frühzeitige und vollständige Beseitigung der Folgen des Gendefekts und damit der Symptome. Beim Typ I gelingt das gut, solange die Behandlung konsequent durchgeführt wird. Auch bei Typ III ist dann die Prognose eher günstig: Es kommt dann nicht zu neurologischen Einschränkungen wie einer Verminderung der Intelligenz. Bei Typ II der Erkrankung droht dagegen oft noch ein tödlicher Ausgang. Dem medizinisch notwendigen Einsatz der Therapie stehen in vielen Ländern die hohen Kosten entgegen.
Literatur
- O. Harmanci, Y. Bayraktar: Gaucher disease: new developments in treatment and etiology. In: World J Gastroenterol., 2008 Jul 7;14(25), S. 3968–3973. Review. PMID 18609679
- J. Schmitz, L. W. Poll, S. vom Dahl: Therapy of adult Gaucher disease. In: Haematologica, 2007 Feb;92(2), S. 148–152. Review. PMID 17296562
Weblinks
Einzelnachweise
- HGMD® gene result. Abgerufen am 21. August 2020.
- OMIM Entry – #230800 – Gaucher Disease, Type I. Abgerufen am 31. Januar 2019 (amerikanisches Englisch).
- OMIM Entry – #231000 – Gaucher Disease, Type III. Abgerufen am 31. Januar 2019 (englisch).
- Th. Stallmach, G. Klöppel, J. Roth, G. A. Spinas: Stoffwechselerkrankungen. In: W. Böcker, H. Denk, Ph. U. Heitz, H. Moch: Pathologie. 4. Auflage. München 2008, S. 1126–1127.
- Gerd Herold u. a.: Innere Medizin. Köln 2009, S. 111.
- Robert J. Hopkin, Gregory A. Grabowski: Lysosomal Storage Diseases. In: Anthony Faucy u. a.: Harrison’s Principles of Internal Medicine. 17. Auflage. New York, 2008, S. 2452–2456.
- The Human Gene Mutation Database. Abgerufen am 31. Januar 2019.
- M. Beck: Volltext-pdf Therapie lysosomaler Speicherkrankheiten. (PDF; 150 kB) In: Deutsches Ärzteblatt, Band 98, Nummer 34–35, S. 2188–2192.
- Gaucher-Krankheit. In: www.symptoma.com. Abgerufen am 7. Dezember 2015.
- Drugs@FDA: FDA Approved Drug Products. Abgerufen am 22. Oktober 2018.
- Eintrag zu Taliglucerase alfa in der DrugBank der University of Alberta, abgerufen am 22. Oktober 2018.
- Pramod K. Mistry, Elena Lukina, Hadhami Ben Turkia, Suma P. Shankar, Hagit Baris: Outcomes after 18 months of eliglustat therapy in treatment-naïve adults with Gaucher disease type 1: The phase 3 ENGAGE trial. In: American Journal of Hematology. Band 92, Nr. 11, 3. Oktober 2017, S. 1170–1176, doi:10.1002/ajh.24877, PMID 28762527, PMC 5656936 (freier Volltext).
- Lesley J. Scott: Eliglustat: A Review in Gaucher Disease Type 1. In: Drugs. Band 75, Nr. 14, September 2015, S. 1669–1678, doi:10.1007/s40265-015-0468-9.
- Elena Lukina, Nora Watman, Marta Dragosky, Heather Lau, Elsa Avila Arreguin: Outcomes after 8 Years of Eliglustat Therapy for Gaucher Disease Type 1: Final Results from the Phase 2 Trial. In: American Journal of Hematology. 28. September 2018, doi:10.1002/ajh.25300.
- Cerdelga®. Abgerufen am 22. Oktober 2018.
- I. Maire, N. Guffon, R. Froissart: [Current development and usefulness of biomarkers for Gaucher disease follow up]. In: La Revue de médecine interne, Band 28 Suppl 2, Oktober 2007, S. S187–S192. PMID 18228687. (Review).