Fries-Umlagerung
Die Fries-Umlagerung (oder Fries-Verschiebung) ist eine Namensreaktion der Organischen Chemie. Die Reaktion ist nach ihrem Entdecker, dem deutschen Chemiker Karl Fries (1875–1962), benannt. Die Fries-Umlagerung beschreibt dabei die elektrophile Umlagerung von Arylestern (beispielsweise Phenylester) unter Lewis-Säure-Katalyse zum entsprechenden Arylketon.[1]
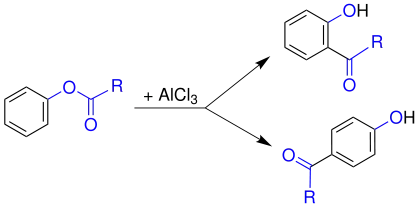
Mechanismus
Trotz erheblicher Anstrengungen konnten noch keine abschließenden Aussagen zum Mechanismus gesichert werden. So wurden in Kreuzexperimenten sowohl Hinweise für intra- als auch intermolekulare Abläufe gefunden. Der Ablauf der Reaktion ist sowohl vom Lösemittel als auch dem Substrat abhängig. Allgemein jedoch wird der untenstehende Mechanismus als bislang beste Erklärung akzeptiert. In einem ersten Schritt greift die Lewis-Säure (hier Aluminiumchlorid, AlCl3) an dem Carbonylsauerstoff der Acylgruppe (1) an. Dieser ist stärker negativiert als der phenolische Sauerstoff und somit bevorzugtes Ziel. Dadurch wird die Bindung des Acylrestes zum phenolischen Sauerstoff polarisiert. Dann wandert das Aluminiumchlorid zum phenolischen Sauerstoff (3). Dabei wird durch Verschiebung der Bindungselektronen ein Acylium-Kation 5 freigesetzt.

Dieses Acylium-Kation 5 reagiert nun unter klassischer elektrophiler Aromatensubstitution mit dem Aromaten 4. Möglich ist ein Angriff in ortho oder para-Stellung. Die Orientierung der Substitution ist temperaturabhängig. Bei niedriger Temperatur wird bevorzugt das para-Produkt, bei hoher Temperatur das ortho-Produkt gebildet. Um den aromatischen Zustand wiederherzustellen, wird nach dem elektrophilen Angriff ein Proton abgespalten und das Keton 7 (ortho-Substitution) bzw. 10 (para-Substitution) erhalten. Das entstandene Keton bleibt als Anion an das Aluminium gebunden. Die Aluminiumverbindung wird anschließend durch Zugabe von Wasser hydrolysiert und das gewünschte Hydroxyketon 8 (ortho-Substitution) bzw. 11 (para-Substitution) wird erhalten.[2][3]
Ortho-Substitution:

Para-Substitution:

Photo-Fries-Umlagerung
Neben der oben beschriebenen Reaktion der Phenylester existiert eine als Photo-Fries-Umlagerung bezeichnete Variante, die über einen radikalischen Mechanismus verläuft. Sie kann auch bei Anwesenheit deaktivierender Gruppen am Aromaten genutzt werden, wird aber aufgrund der in der Regel schlechten Ausbeuten bislang nur im Labor eingesetzt. Wenn die para-Position am Phenylrest durch einen Substituenten (z. B. eine Methylgruppe) blockiert ist, entstehen nur ortho-Hydroxyketone.[4] Ist der Phenylrest in para-Position nicht substituiert, entstehen Gemische der ortho- und para-Hydroxyketone.[5] Der erste Schritt des Mechanismus beschreibt die Bildung eines Acylradikals 13 und eines Aryloxiradikals 12, das sich durch seine mesomere Stabilisierung auszeichnet.

Die mesomeren Grenzstrukturen geben dabei vor, dass das entstandene Acylradikal 13 entweder in ortho- oder in para-Stellung an das Aryloxiradikals 12 binden kann. Nach der Umlagerung eines Protons entsteht dann das entsprechende Hydroxyketon 8 bzw. 11.[3]
Ortho-Verschiebung

Para-Verschiebung

Bedeutung
Da die Reaktion von Phenolen mit Acylhalogeniden unter den Bedingungen der Friedel-Crafts-Acylierung Phenylester, nicht aber die gewünschten Hydroxyarylketone liefert, ist die Reaktion von großtechnischer Bedeutung für die Synthese der Hydroxyarylketone die als wichtige Ausgangsstoffe für die Synthese verschiedener Pharmazeutika (z. B. Paracetamol oder Salbutamol) dienen. Anstelle des Aluminiumchlorids können zum Teil auch andere Lewissäuren (Bortrifluorid, Bismuttriflat etc.) oder auch starke protische Säuren (Flusssäure oder Methansulfonsäure) zum Einsatz kommen. Um den Verbrauch dieser korrosiven und ökologisch bedenklichen Katalysatoren zu vermeiden, wird intensiv der Einsatz von Festkörperkatalysatoren untersucht.
Die photochemische Fries-Umlagerung ist ein unerwünschter Effekt bei der lichtinduzierten Verfärbung von an sich farblosen Kunststoffen. Dem kann durch Einbau von geeigneten Radikalfängern entgegengewirkt werden.
Grenzen
In allen Fällen können nur solche Ester verwendet werden deren Acylkomponente unter den harschen Bedingungen stabil ist. Ist der Aromat oder die Acylkomponente hochsubstituiert, sinkt aufgrund zu starker sterischer Beanspruchung die Ausbeute stark ab. Deaktivierende (meta-dirigierende) Gruppen am Aromaten lassen die Ausbeuten drastisch absinken, wie für eine Friedel-Crafts-Reaktion zu erwarten ist.
Literatur
- K. Fries, G. Finck: Über Homologe des Cumaranons und ihre Abkömmlinge. In: Berichte der deutschen chemischen Gesellschaft. Band 41, Nr. 3, 1. Oktober 1908, S. 4271–4284, doi:10.1002/cber.190804103146.
- K. Fries, G. Finck: Über Sauerstoff-Isologe homologer Indirubine. In: Berichte der deutschen chemischen Gesellschaft. Band 41, Nr. 3, 1. Oktober 1908, S. 4284–4294, doi:10.1002/cber.190804103147.
- K. Fries, W. Pfaffendorf: Über ein Kondensationsprodukt des Cumaranons und seine Umwandlung in Oxindirubin. In: Berichte der deutschen chemischen Gesellschaft. Band 43, Nr. 1, 1. Januar 1910, S. 212–219, doi:10.1002/cber.19100430131.
- Jerry March: Advanced Organic Chemistry. 3rd Ed., John Wiley & Sons, 1985, ISBN 0-471-88841-9, S. 499–500.
Weblinks
- Fries Rearrangement. www.organic-chemistry.org, abgerufen am 9. März 2009 (englisch).
Einzelnachweise
- Louis Fieser, Mary Fieser: Organische Chemie. 2. Auflage, Verlag Chemie Weinheim, 1972, ISBN 3-527-25075-1, S. 926–928.
- Thomas Laue, Andreas Plagens: Namens- und Schlagwort-Reaktionen der Organischen Chemie, 5. Auflage, Teubner Studienbücher Chemie, 2006, S. 124.
- Zerong Wang: Comprehensive Organic Name Reactions and Reagent (3-Volume Set) Volume 1, Wiley, 2009, ISBN 978-0-471-70450-8, S. 1143.
- Jürgen Martens, Klaus Praefcke: Organische Schwefelverbindungen, VII. Photochemische α-Spaltung von Thiobenzoesäure-S-p-tolylestern in Lösung. In: Chemische Berichte. Band 107, Nr. 7, 1. Juli 1974, S. 2319–2325, doi:10.1002/cber.19741070716.
- Louis Fieser, Mary Fieser: Organische Chemie. 2. Auflage, Verlag Chemie Weinheim, 1972, ISBN 3-527-25075-1, S. 928–929.