Erbkrankheit
Als Erbkrankheit (oder genetisch bedingte Krankheit) werden Erkrankungen und Besonderheiten bezeichnet, die entweder durch eine Mutation (Genvariante) in einem Gen (monogen) oder durch mehrere Mutationen (Genvarianten) in verschiedenen Genen (polygen) ausgelöst werden können und die zu bestimmten Erkrankungsdispositionen führen. In diesem Zusammenhang spricht man auch von monogenetischer bzw. polygenetischer Erkrankung.
- Beispiele für Erbgänge
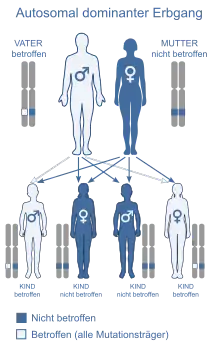 Autosomal-dominanter Erbgang
Autosomal-dominanter Erbgang Autosomal-rezessiver Erbgang
Autosomal-rezessiver Erbgang
Im engeren Sinne zählt man jedoch nur jene Erkrankungen und Besonderheiten zu den Erbkrankheiten, die durch von Anfang an untypisch veränderte Gene ausgelöst und durch Vererbung von den Vorfahren auf ihre Nachkommen übertragen werden. Die früheste Methode zur Erforschung der Vererbungswege war die Stammbaumanalyse bei Familienstammbäumen, in denen beispielsweise die Bluterkrankheit oder die Farbenblindheit usw. gehäuft auftraten.[1]
Syndrome wie Formen von Trisomie, bei denen sich nicht die übliche Zahl von 46 Chromosomen im menschlichen Genom findet, können somit genau genommen nicht als Erbkrankheit gezählt werden, da sie zumeist spontan erst bei der Zellteilung des Embryos auftreten und daher selten von einem Elternteil geerbt werden.
Verschiedene Formen
Erbkrankheiten folgen verschiedenen Erbgängen und sind mit unterschiedlichen Vererbungs-, Wiederholungs- und Erkrankungswahrscheinlichkeiten verbunden. Man unterscheidet autosomal-rezessive und autosomal-dominante von gonosomalen und mitochondrialen Erbgängen.
Autosomal-rezessive Erbgänge

Autosomal-rezessive Erbkrankheiten sind geschlechterunabhängig. Die Besonderheit tritt nur dann in Erscheinung, wenn sich auf jeweils beiden Chromosomen eine Veränderung (Mutation) in beiden Kopien eines bestimmten Gens findet, d. h., wenn der betreffende Mensch jeweils eine Veränderung von seinem biologischen Vater und eine von seiner biologischen Mutter geerbt hat. Die Eltern müssen dabei nicht betroffen sein, der Phänotyp tritt also nicht in jeder Generation auf. Die Mutation muss dabei nicht identisch sein. Führen zwei molekulargenetisch unterscheidbare Mutationen zu dem gleichen Funktionsverlust in einem Gen, so spricht man von Compound Heterozygotie. Beispiele für autosomal-rezessive Erbgänge sind Mukoviszidose, Albinismus und Phenylketonurie (PKU) (Defekt der Phenylalaninhydroxylase).
Bei autosomal-rezessiv vererbten Erkrankungen handelt es sich meist um Loss-of-Function-Mutationen (Funktionsverlustmutationen). Ursachen für scheinbare Abweichungen autosomal-rezessiver Vererbung sind Pseudodominanz, Heterogenie, Isodisomie und das Nichteinrechnen von Heterozygoten mit kranken Kindern. Typische Beispiele sind:
- Adrenogenitales Syndrom (AGS)
- Ahornsirupkrankheit
- Albinismus
- Alkaptonurie
- Alpha1-Antitrypsinmangel
- Autosomal-rezessive polyzystische Nierenerkrankung (ARPKD)
- Galaktosämie
- Hereditäre Fruktoseintoleranz
- Hämochromatose
- Joubert-Syndrom
- Kombinierte Malon- und Methylmalonazidurie (CMAMMA)
- Kretinismus
- Kurzripp-Polydaktylie-Syndrome (Typ I, II, III, IV)
- Laurence-Moon-Biedl-Bardet-Syndrom (LMBB-Syndrom)
- Lippen-Kiefer-Gaumenspalte
- Morbus Wilson
- Mukopolysaccharidosen (MPS)
- Mukoviszidose bzw. Zystische Fibrose
- Nephrotisches Syndrom vom finnischen Typ
- Peters-Plus-Syndrom
- Phenylketonurie (PKU)
- Ribbing-Syndrom
- Thalassämie
- Xeroderma pigmentosum
Autosomal-dominante Erbgänge

Hier führt bereits ein verändertes Allel (Allele sind die einander jeweils und gleichzeitig gegensätzlich entsprechenden Gene eines diploiden Chromosomensatzes) auf einem der beiden homologen Chromosomen zur Merkmalsausprägung. Die genetische Information liegt auf einem der 44 Autosomen vor und wird unabhängig vom Geschlecht vererbt. Frauen und Männer sind also gleichermaßen betroffen. Der Phänotyp tritt in jeder Generation auf. Beispiele sind:
- Achondroplasie
- Apert-Syndrom
- Autosomal-dominante polyzystische Nierenerkrankung (ADPKD)
- Brachydaktylie
- Chorea Huntington („Veitstanz“)
- Ehlers-Danlos-Syndrom (Typen I–IV, VII A/B, VIII)
- Engelmann-Syndrom
- Erythropoetische Protoporphyrie
- Faktor-V-Leiden-Mutation
- Familiäre Hypercholesterinämie
- HMSN Typ I (Morbus Charcot-Marie-Tooth)
- Maligne Hyperthermie
- Marfan-Syndrom
- Morbus Darier
- Multiple kartilaginäre Exostosen
- Myotone Dystrophie Typ I
- Neurofibromatose (Morbus Recklinghausen)
- Okulopharyngeale Muskeldystrophie (OPMD)
- Osteogenesis imperfecta (Typ I)
- Piebaldismus
- Polydaktylie
- Retinoblastom
- Ruvalcaba-Myhre-Smith-Syndrom und
- Sichelzellenanämie
- Sotos-Syndrom
- Tuberöse Sklerose
Gonosomale Erbgänge
Gonosomale Erbkrankheiten, also solche, bei denen die Veränderung die Geschlechtschromosomen X bzw. Y betrifft, liegen in den meisten Fällen auf dem X-Chromosom, da das Y-Chromosom weniger Gene enthält. Das X-Chromosom hat 155 Megabasen, das Y-Chromosom 59 Megabasen.[2] Am Beispiel der X-chromosomalen Vererbung werden folgende Besonderheiten deutlich:
X-chromosomal-rezessiv
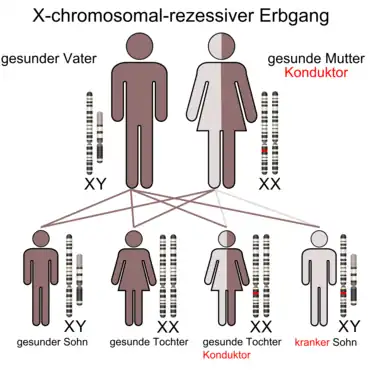 X-chromosomal-rezessiver Erbgang (Mutter ist Konduktor)
X-chromosomal-rezessiver Erbgang (Mutter ist Konduktor) X-chromosomal-rezessiver Erbgang (bei krankem Vater)
X-chromosomal-rezessiver Erbgang (bei krankem Vater)
Mädchen/Frauen sind nur betroffen, wenn beide X-Chromosomen geschädigt sind, ansonsten sind sie nur Anlageträger (Konduktoren), d. h., sie können das veränderte X-Chromosom an ihre Kinder weitervererben, bilden selbst aber keinen entsprechenden Phänotyp aus. Mädchen/Frauen können vielfach die Veränderung auf einem X-Chromosom durch ihr zweites X-Chromosom ausgleichen, wenn es nicht verändert ist. Jungen/Männer sind dann betroffen, wenn sie das eine veränderte X-Chromosom von der phänotypisch gesunden Mutter, oder eines von beiden veränderten X-Chromosomen einer phänotypisch erkrankten Mutter vererbt bekommen, da Jungen/Männer ja ein X-Chromosom auf jeden Fall von der Mutter bekommen und auch nur dieses eine besitzen. Phänotypisch sind Jungen/Männer also häufiger betroffen, da Mädchen/Frauen den Defekt durch das andere X-Chromosom ausgleichen. Beispiele sind Glucose-6-Phosphat-Dehydrogenase-Mangel (G-6-PD-Mangel), Hämophilie A und B (Bluterkrankheit), Lesch-Nyhan-Syndrom, Morbus Fabry, Mukopolysaccharidose Typ II, Muskeldystrophie (Typ Duchenne, Typ Becker-Kiener), Norrie-Syndrom, Retinitis pigmentosa, Rot-Grün-Blindheit, Septische Granulomatose, X-SCID (severe combined immune deficiency) und Ornithin-Transcarbamylase (OTC)-Mangel[3] (Harnstoffzyklusdefekt).
X-chromosomal-dominant
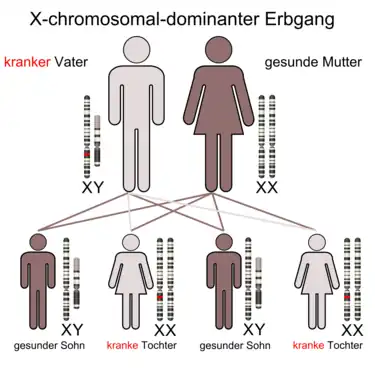 X-chromosomal-dominanter Erbgang (bei krankem Vater)
X-chromosomal-dominanter Erbgang (bei krankem Vater) X-chromosomal-dominanter Erbgang (bei kranker Mutter)
X-chromosomal-dominanter Erbgang (bei kranker Mutter)
Jungen/Männer sind zu 50 % betroffen, wenn ihre Mutter Trägerin eines auf einem X-Chromosoms liegenden krankmachenden Allels ist. Enthalten dagegen ihre beiden X-Chromosomen das krankmachende Allel, so sind alle Kinder betroffen. Mädchen/Frauen sind insgesamt häufiger betroffen, da die Wahrscheinlichkeit, ein verändertes X-Chromosom zu erhalten, bei zwei X-Chromosomen (eins vom Vater, eins von der Mutter) höher ist als bei Jungen/Männern (Eines von der Mutter). Beispiele sind Familiäre phosphatämische Rachitis (auch idiopathisches Debré-de-Toni-Fanconi-Syndrom oder Vitamin-D-resistente Rachitis genannt), Rett-Syndrom und Oro-fazio-digitales Syndrom Typ 1.
Mitochondriale bzw. Extrachromosomale Erbgänge
Etwa 0,1 Prozent der DNA einer menschlichen Zelle befinden sich nicht im Zellkern, sondern in den Mitochondrien. Da Eizellen im Gegensatz zu Spermien mehrere hunderttausend Mitochondrien besitzen, werden Mutationen in der Mitochondrien-DNA nur mütterlicherseits vererbt. Gleiches gilt für die Chloroplasten photosynthetisch aktiver Organismen.
Siehe auch Extrachromosomale Vererbung
Diagnose und Behandlung
| Klassifikation nach ICD-10 | |
|---|---|
| Q90 - Q99 | Chromosomenanomalien, anderenorts nicht klassifiziert |
| ICD-10 online (WHO-Version 2019) | |
Bei Verdacht auf eine Erbkrankheit kann eine humangenetische Untersuchung Klarheit verschaffen. Dabei werden die Chromosomen auf zahlenmäßige und strukturelle Veränderungen überprüft. Besteht dringender Verdacht auf einen bestimmten genetischen Defekt ist auch eine weitergehende, aufwändige Untersuchung einzelner Genkonstellationen möglich. Die Ergebnisse können dann bei der Risikoabschätzung bzgl. einer Vererbung hilfreich sein.
Therapeutisch kann bei einer vorliegenden Besonderheit des Erbguts mit den heutigen medizinischen Möglichkeiten meist nicht auf die Ursachen eingewirkt werden. Es werden daher meist Ratschläge in Bezug auf die Lebensweise, Aufklärung über Risikofaktoren und symptomatische Maßnahmen getroffen. Dies sind dann individuelle Entscheidungen, zumal es sich nicht immer um eine Krankheit, sondern oft um eine Disposition handelt.
Für einige wenige Erkrankungen, wie z. B. die Spinale Muskelatrophie gibt es erste Therapieversuche.
Geschichte
Der erst seit dem 20. Jahrhundert in der Bedeutung genetische Krankheit verwendete Begriff der Erbkrankheit[4] wurde in der ersten Hälfte des 20. Jahrhunderts auch häufig falsch verwendet, unter anderem für angebliche „Krankheiten“ wie „kriminelle Neigung“ oder „Asozialität“.[5] Dieses Denken beeinflusste Sterilisations-Programme[6] und den Euthanasie-Gedanken und fand seine extreme Ausprägung im deutschen Nationalsozialismus, war aber zum damaligen Zeitpunkt auch in vielen anderen Ländern wie den USA, England und Frankreich vorhanden. Heute werden nur noch solche Krankheiten als Erbkrankheiten bezeichnet, die möglichst klar abgrenzbar sind und mit sehr hoher Wahrscheinlichkeit auf Gendefekte zurückgehen.
Sonstige Erbkrankheiten und Besonderheiten
Genetisch bedingte Disposition
Diverse Erkrankungen, Behinderungen und Besonderheiten sind nicht im Sinne einer klassischen Erbkrankheit erblich, sondern ihr Auftreten kann durch eine (mitunter familiäre) genetische Erkrankungsdisposition (Veranlagung, Anfälligkeit) bedingt sein. Hierzu zählen z. B.:
- Adipositas
- Allergien, diverse
- Alzheimer-Krankheit
- Autoimmunerkrankungen
- Bipolare Störung
- Bluthochdruck
- Creutzfeldt-Jakob-Krankheit
- Depression
- Diabetes mellitus
- Hallux valgus
- Haarausfall
- Herzfehler
- Herzinfarkt
- Krebserkrankungen diverse (siehe Richtlinien zur Diagnostik der genetischen Disposition für Krebserkrankungen auf der Website der Bundesärztekammer)
- Laktoseintoleranz
- maligne Hyperthermie
- Migräne
- Multiple Sklerose (MS)
- Osteoporose
- Parkinson-Krankheit
- Psoriasis
- Rheuma
- Schizophrenie
- Schlaganfall
- Taubheit
- Formen der Trisomie (Disposition zur Entstehung einer Translokations-Trisomie bei Nachkommen beim Vorliegen einer „Balancierten Translokation“ des entsprechenden Chromosoms bei Eltern ohne die jeweilige Form von Trisomie)
- Vitiligo
Siehe auch
- Liste von Erbkrankheiten
- Genetik
- Erbliche Tumorerkrankungen
- Pränataldiagnostik
- Präimplantationsdiagnostik
- Erbkrankheiten der Hunde (Kategorie)
Weblinks
Einzelnachweise
- Ulrich Weber: Biologie Oberstufe. Gesamtband. Cornelsen, Berlin 2001, ISBN 3-464-04279-0, S. 180–182.
- Ensembl Datenbank, abgerufen am 11. Februar 2017.
- J. E. Wraith: Ornithine carbamoyltransferase deficiency. In: Archives of Disease in Childhood. Januar 2001, Band 84, Nr. 1, S. 84–88: Review. PMID 11124797.
- Werner Sohn: Erbkrankheiten. In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin/ New York 2005, ISBN 3-11-015714-4, S. 366 f.; hier: S. 366.
- Wolfgang Ayaß: „Asozialer Nachwuchs ist für die Volksgemeinschaft vollkommen unerwünscht“. Die Zwangssterilisationen von sozialen Außenseitern. In: Margret Hamm (Hrsg.): Lebensunwert – zerstörte Leben. Zwangssterilisation und „Euthanasie“. Verlag für akademische Schriften (VAS), Frankfurt am Main 2005, ISBN 3-88864-391-0, S. 111–119.
- Vgl. etwa Fred Nöller: Chirurgisch-orthopädische Erbkrankheiten im Gesetz zur Verhütung erbkranken Nachwuchses. G. Fischer, Jena 1942.