Dinitromethan
Dinitromethan ist der zweifachsubstituierte Vertreter der Reihe der Nitromethane mit Nitromethan, Dinitromethan, Trinitromethan und Tetranitromethan sowie die einfachste geminale Dinitroalkylverbindung. Die Verbindung ist oberhalb von Raumtemperatur zunehmend instabil. Eine sichere Handhabung erfolgt eher in Form ihrer Alkalisalze.
| Strukturformel | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||
| Allgemeines | |||||||||||||
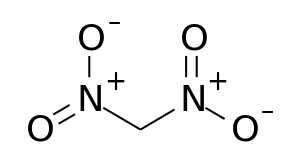
| Name | Dinitromethan | ||||||||||||
| Summenformel | CH2N2O4 | ||||||||||||
| Kurzbeschreibung |
farblose Flüssigkeit mit schwachem angenehmen Geruch[1] | ||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||
| |||||||||||||
| Eigenschaften | |||||||||||||
| Molare Masse | 106,0376 g·mol−1 | ||||||||||||
| Aggregatzustand |
flüssig | ||||||||||||
| Dichte |
1,524 g·cm−3 (20 °C)[1] | ||||||||||||
| Siedepunkt | |||||||||||||
| pKS-Wert |
3,57 (20 °C)[2] | ||||||||||||
| Brechungsindex |
1,4480 (20 °C)[1] | ||||||||||||
| Sicherheitshinweise | |||||||||||||
| |||||||||||||
| Thermodynamische Eigenschaften | |||||||||||||
| ΔHf0 |
−104,9 kJ/mol[4] | ||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Brechungsindex: Na-D-Linie, 20 °C | |||||||||||||
Geschichte
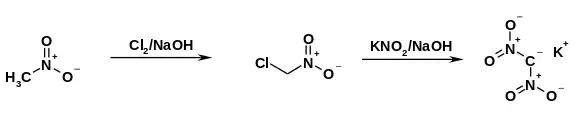
Eine erste, unsichere Quelle der Herstellung von Dinitromethan stammt aus dem Jahr 1878, als Dinitromethan in heftiger Reaktion aus Aceton und konzentrierter Salpetersäure entstanden sein soll.[5][6] Die ersten Herstellungen des Kaliumsalzes gelangen A. Villiers 1884[7] und Paul Duden 1893[6] in geringen Ausbeuten aus Bromdinitromethan, das durch Erhitzen von 2,4,6-Tribromanilin mit konzentrierter Salpetersäure erhalten wurde. Eine 1951 von Feuer et al.[8] veröffentlichte zweistufige Synthesevariante geht von Nitromethan aus, das zunächst zum Chlornitromethan chloriert wird. Durch nukleophile Substitution mittels Kaliumnitrit im basischen Medium entsteht das Kaliumsalz. Die Ausbeute dieser Variante liegt allerdings nur bei 23 %.
Die Einwirkung von konzentrierten oder mäßig verdünnten Säuren auf das Kaliumsalz führte bei Raumtemperatur zur Zersetzung unter lebhafter Bildung nitroser Gase. Eine Freisetzung des Dinitromethans gelingt nur in der Kälte in etherischer Lösung durch vorsichtige, portionsweise Zugabe verdünnter Schwefelsäure.[6]
Darstellung und Gewinnung
Eine Synthese geht vom Malonsäuremonomethylester aus, der durch oxidative Nitrierung unter Decarboxylierung in 2,2-Dinitroessigsäuremethylester umgewandelt wird. Der zweite Decarboxylierungsschritt erfolgt bei der basischen Hydrolyse dieses Esters mittels Natronlauge, wobei zunächst das Natriumsalz des Dinitromethans entsteht.[9]

Eine neuere Synthese startet mit der Nitrierung von Barbitursäure unter milden Bedingungen. Die resultierende 5,5-Dinitrobarbitursäure kann schon durch Wasser nucleophil hydrolysiert werden, wobei unter Abspaltung von Kohlendioxid 2,2-Dinitroacetylharnstoff entsteht. Dieser kann in der Hitze mit Kalilauge zum Kaliumsalz von Dinitromethan und Harnstoff zersetzt werden. Die Gesamtausbeute dieser Synthesevariante liegt bei 80 %.[10]

Dinitromethan ist auch ein Nebenprodukt der Synthese von Diaminodinitroethylen. Die Synthese geht vom 2,6-Dihydroxy-4-methylpyrimidin aus, das durch Nitrierung in Nitriersäure zu einem Tetranitrozwischenprodukt umgesetzt wird. Dieses wird danach hydrolytisch zu Dinitromethan, Diaminodinitroethylen und Kohlenstoffdioxid gespalten.[11]
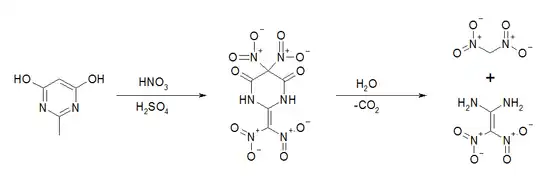
Diaminodinitroethylen besitzt acide Eigenschaften. In Gegenwart von Basen erfolgt eine Deprotonierung. Durch Umsetzung mit Kalilauge bei niedrigen Temperaturen lässt sich das Kaliumsalz als weißer, kristalliner Feststoff isolieren. Erhitzen auf 70 °C mit Kalilauge führt zu einer basischen Hydrolyse, wobei das Kaliumsalz des Dinitromethans und Harnstoff gebildet werden.[12]

Die Freisetzung von Dinitromethan aus dem Kaliumsalz kann durch das vorsichtige Einleiten von Fluorwasserstoff in eine etherische Lösung des Salzes bei 0–5 °C erfolgen.[1]
Eigenschaften
Dinitromethan ist je nach Reinheit ein farbloses bis gelbliches Öl.[6][1] Die Verbindung ist instabil und zersetzt sich langsam bei Raumtemperatur.[6] Bei 0 °C ist die Verbindung über Monate stabil.[1] Eine Destillation wurde im Vakuum bei 2 Torr mit einem Siedepunkt bei 39–40 °C bzw. bei 4 Torr bei 52–53,5 °C durchgeführt.[1] Wegen der Fähigkeit zum explosionsartigen Zerfall wird davon aber abgeraten. Für die Verbindung wurde eine Detonationswärme von 6814 kJ·mol−1 abgeschätzt.[13][14] Durch den −M-Effekt der Nitrogruppen ist Dinitromethan C-H-acid und mit einem pKs-Wert von 3,57 eine mittelstarke Säure.[2] Seine Säurestärke ist vergleichbar mit der von Ameisensäure (pKs 3,75). Für Dinitromethan können zwei tautomere Strukturen formuliert werden. Das Gleichgewicht liegt auf der Seite der C-H-aciden Struktur. Quantenchemische Berechnungen ergeben eine Differenz der freien Enthalpie von 19,6 kJ·mol−1 zur N-OH-aciden Struktur.[15] Das 1H-NMR-Spektrum zeigt nur ein einziges Signal bei 3,9 ppm für die C-H-Funktion.[16] Erwartungsgemäß liegt dieser Wert zwischen denen für Nitromethan mit 5,72 ppm und Trinitromethan mit 2,48 ppm.[16] Der Vergleich quantenchemisch berechneter und experimenteller IR- und Raman-Spektren bestätigt die C-H-acide Struktur.[17]

In stark saurem Medium kann das Gleichgewicht stabilisiert durch ein Protolysegleichgewicht in Richtung der N-OH-aciden Struktur verschoben werden.[18][19] Durch Eintragen des Kaliumsalzes von Dinitromethan in konzentrierte oder rauchende Schwefelsäure kann das Cyclodimerisierungsprodukt Dinitrofuroxan erhalten werden. Die Bildung dieses Dimers verläuft über die Intermediate der protonierten N-OH-aciden Struktur des Dinitromethans und des daraus durch Wasserabspaltung resultierenden Nitroformonitriloxids.[19]

Die Salze des Dinitromethans zeichnen sich durch signifikant höhere Stabilität im Vergleich zur freien Verbindung aus. So wird beim Kaliumsalz erst ab 200 °C eine explosionsartige Zersetzung beobachtet.[8] Als Zersetzungsprodukte wurden Kaliumcarbonat, Wasser, Kohlendioxid, Stickstoffmonoxid und Stickstoff detektiert.[6] Die Salze organischer Basen beginnen sich um 100 °C zu zersetzen. So wurde für das Piperaziniumsalz eine Zersetzung ab 90 °C, für das Formamidiniumsalz ab 125 °C und das Guanidiniumsalz ab 175 °C beobachtet.[20]
Bei chemischen Umsetzungen geht man meist vom Kalium- oder Natriumsalz des Dinitromethans aus. Das Anion wirkt als gutes Nucleophil. So reagiert es mit Formaldehyd zum 2,2-Dinitropropan-1,3-diol.[8]

Die Reaktion mit Dialkyl- oder Diarylsulfoxiden ergibt die entsprechenden Sulfanylidendinitromethane.[21][22]

Verwendung
Trotz der Explosionsfähigkeit wird die Verbindung wegen ihrer schlechten Handhabbarkeit nicht als Explosivstoff verwendet. Die Salze des Dinitromethans können als Synthesebausteine für Heterocyclensynthesen eingesetzt werden. Die Salze mit alkylierten Imidazolen können als ionische Flüssigkeit verwendet werden.[23]
Einzelnachweise
- Legin, G. Ya.; Okhlobystina, L. V.; Fainzilberg, A. A.: Preparation of pure dinitromethane and its properties. In: Russian Chemical Bulletin. 14. Jahrgang, Nr. 12, 1965, S. 2190–2191, doi:10.1007/BF00846018.
- Adolph, H. G.; Kamlet, M. J.: Fluoronitroaliphatics. I. The Effect of α Fluorine on the Acidities of Substituted Nitromethanes. In: Journal of the American Chemical Society. 88. Jahrgang, Nr. 20, 1966, S. 4761–4763, doi:10.1021/ja00972a065.
- Dieser Stoff wurde in Bezug auf seine Gefährlichkeit entweder noch nicht eingestuft oder eine verlässliche und zitierfähige Quelle hierzu wurde noch nicht gefunden.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press / Taylor and Francis, Boca Raton FL, Standard Thermodynamic Properties of Chemical Substances, S. 5-19.
- Chancel: Compt. Rend. 86 (1878) 1405 und Jahresbericht für 1878, 694
- Duden, P.: Ueber das Dinitromethan. In: Berichte der Deutschen Chemischen Gesellschaft. 26. Jahrgang, Nr. 3, 1893, S. 3003–3011, doi:10.1002/cber.189302603135.
- A. Villiers: Bull. Soc. Chim. Fr. 41 (1884) 281 und 43 (1886) 322.
- Feuer, H.; Bachmann, G. B.; Kispersky, J. P.: A New Preparation of Potassium Dinitromethane and its Conversion to 2,2-Dinitro-1,3-propanediol. In: Journal of the American Chemical Society. 73. Jahrgang, Nr. 3, 1951, S. 1360, doi:10.1021/ja01147a511.
- Grakauskas, V.; Guest, A. M.: Dinitromethane. In: Journal of Organic Chemistry. 43. Jahrgang, Nr. 18, 1978, S. 3485–3488, doi:10.1021/jo00412a014.
- Langlet, A.; Latypov, N. V.; Wellmar, U.; Goede, P.; Bergman, J.: Synthesis and reactions of 5,5-dinitrobarbituric acid. In: Tetrahedron Letters. 41. Jahrgang, Nr. 12, 2000, S. 2011–2013, doi:10.1016/S0040-4039(00)00086-1.
- Latypov, N. V.; Johansson, M.; Holmgren, E.; Sizova, E. V.; Sizov, V. V.; Bellamy, A. J.: On the Synthesis of 1,1-Diamino-2,2-dinitroethene (FOX-7) by Nitration of 4,6-Dihydroxy-2-methylpyrimidine. In: Organic Process Research and Development. 11. Jahrgang, Nr. 1, 2007, S. 56–59, doi:10.1021/op068010t.
- Bellamy, A. J.: FOX-7 (1,1-Diamino-2,2-dinitroethene). In: Structure and Bonding. 125. Jahrgang, 2007, S. 1–33, doi:10.1007/430_2006_054.
- Zeman, S.: New application of kinetic data of the low-temperature thermolysis of nitroparaffins. In: Thermochimica Acta. 261. Jahrgang, 1995, S. 195–207, doi:10.1016/0040-6031(95)02325-V.
- Zeman, S.: Modified Evans-Polanyi-Semenow relationship in the study of chemical micromechanism governing detonation initiation of individual energetic materials. In: Thermochimica Acta. 384. Jahrgang, Nr. 1–2, 2002, S. 137–154, doi:10.1016/S0040-6031(01)00787-0.
- Brand, H.; Liebman, J. F.; Schulz, A.: Cyano-, Nitro- and Nitrosomethane Derivatives: Structures and Gas-Phase Acidities. In: European Journal of Organic Chemistry. 2008. Jahrgang, Nr. 27, 2008, S. 4665–4675, doi:10.1002/ejoc.200800583.
- Hofmann, W.; Stefaniak, L.; Urbanski, T.; Witanowski, M.: Proton Magnetic Resonance Study of Nitroalkanes. In: Journal of the American Chemical Society. 86. Jahrgang, Nr. 4, 1964, S. 554–558, doi:10.1021/ja01058a005.
- Tafipolsky, M. A.; Tokmakov, I. V.; Shlyapochnikov, V. A.: Structure and vibrational spectra of dinitromethane and trinitromethane. In: Journal of Molecular Structure. 510. Jahrgang, Nr. 1–3, 1999, S. 149–156, doi:10.1016/S0022-2860(99)00080-0.
- Edwards, J. T.; Tremaine, P. H.: The Meyer Reaction of Phenylnitromethane in Acid. III. The Tautomerization to the aci-Form. In: Canadian Journal of Chemistry. 49 (21), 1971, S. 3493–3501, doi:10.1139/v71-584.
- Ovchinnikov, I. V.; Makhova, N. N.; Khmelnitskii, L. I.: Nitroformonitril oxide 2. Generation of nitroformonitrile oxide as an intermediate for the preparation of dinitrofuroxan. In: Russian Chemical Bulletin. 44. Jahrgang, Nr. 4, 1995, S. 702–706, doi:10.1007/BF00698507.
- Jalový, Z.; Ottis, J.; Růžička, A.; Lyčka, A.; Latypov, N. V.: Organic salts of dinitromethane. In: Tetrahedron. 65. Jahrgang, Nr. 34, 2009, S. 7163–7170, doi:10.1016/j.tet.2009.06.014.
- Shevelev, S.A. et al. in Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1976, Vol. 25, p. 1906–1909.
- Shitov,O.P. et al. in Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1977, Vol. 26, p. 214–217.
- Brand, H.; Liebman, J. L.; Schulz, A.; Mayer, P.; Villinger, A.: Nonlinear, Resonance-Stabilized Pseudohalogenides: From Alkali Methanides to Ionic Liquids of Methanides. In: European Journal of Inorganic Chemistry. 2006. Jahrgang, Nr. 21, 2006, S. 4294–4308, doi:10.1002/ejic.200600668.