Zerebrale Amyloidangiopathie
Die zerebrale Amyloidangiopathie (auch cerebrale Amyloidangiopathie, CAA) ist eine Erkrankung der Blutgefäße des Gehirns. Es kommt zur Ablagerung von Beta-Amyloid in den Wänden der Blutgefäße, was zur Einengung des Lumens sowie zur Bildung von Mikroaneurysmen führen kann. Diese wiederum können brechen und zu intrazerebralen Blutungen führen.
| Klassifikation nach ICD-10 | |
|---|---|
| I68.0* | Zerebrale Amyloidangiopathie |
| ICD-10 online (WHO-Version 2019) | |
Geschichte
Erstmals beschrieben wurde die zerebrale Amyloidangiopathie in Gehirnen älterer Menschen 1938 von W. Scholz.[1] Aufgrund der Gemeinsamkeit der Ablagerung von Amyloid wurde die CAA ursprünglich als ein Merkmal der Alzheimer-Krankheit gesehen.[2] In den 1970er Jahren konnte man zeigen, dass bei vielen intrazerebralen Blutungen, die nicht auf zu hohen Blutdruck zurückzuführen sind, derartig veränderte Gefäßwände eine Rolle spielen.[3]
Einteilung
Die häufigste Form ist die Beta-Amyloid-Ablagerung, die in bis zu 80 % der Alzheimer-Krankheitsfälle beobachtet wird. Daneben existieren verschiedene erbliche Formen mit Mutationen des Amyloid-Precursor-Proteins (Dutch type), Presenilin, Cystatin-C-Mutation (ACys, Icelandic type) sowie die hereditäre Demenz vom britischen/dänischen Typ.
Nach der medizinischen Datenbank Orphanet können folgende Formen unterschieden werden:
Pathogenese
Beta-Amyloid entsteht durch Zerschneiden des Amyloid-Precursor-Proteins (APP) mithilfe der Enzyme Beta- und Gamma-Sekretase. Im normalen Stoffwechsel werden diese Peptide nicht erzeugt, man nimmt an, dass das Amyloid vorwiegend in Nervenzellen erzeugt wird. Bei Betroffenen reichert es sich im Nervenwasser an und kann sich im Gehirngewebe als sogenannte senile Plaques ablagern, wie etwa bei Alzheimer-Demenz, oder aber in den Gefäßwänden, wie bei der CAA. Dabei wird Amyloid vor allem in der mittleren Gefäßwandschicht (Media) eingelagert.[3] Das Vorliegen des ApoE4-Allels geht mit einem erhöhten Risiko für die Ablagerung von Amyloid in den Gefäßwänden einher.
Diagnostik

Das Vorliegen einer zerebralen Amyloidangiopathie kann nur mittels Obduktion oder durch eine Gewebebiopsie des Gehirns definitiv gesichert werden.[12][13] Die Histologie zeigt typischerweise hyalinisierte Kapillaren mit Nachweis von Beta-Amyloid in den Gefäßen, oft mit Verlust der glatten Muskelzellen einhergehend.
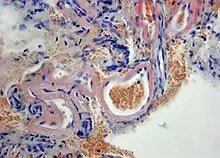 Histopathologie der zerebralen Amyloidangiopathie mit Ablagerung von Amyloid (rot) in der Wand hyalinisierter meningealer Gefäße. Kongorot-Färbung einer Hirnbiopsie. |
 Typische apfelgrüne Doppelbrechung im polarisierten Licht |
Klinisch stützt sich die Diagnose auf das radiologische Vorliegen von einzelnen oder multiplen lobären Blutungen an der Rinden-Mark-Grenze oder von Mikroblutungen im Gehirn ohne andere wahrscheinlichere Ursache.[3][14] Das Vorliegen einer zerebralen Amyloidangiopathie wird als wahrscheinlich angesehen, wenn in der Vorgeschichte mindestens zwei Blutungen ohne andere erkennbare Ursache vorliegen. Man geht davon aus, dass zwischen 5 und 12 % aller intrakraniellen Blutungen bei Patienten über 55 Jahren durch eine zerebrale Amyloidangiopathie verursacht sind.
Einzelnachweise und Quellen
- W. Scholz: Studien zur Pathologie der Hirngefäße. II Die drusige Entartung der Hirnarterien und Kapillaren. In: Z Neurol Psychiatr. Nummer 162, 1938, S. 694–715.
- T. Revesz, J. L. Holton, T. Lashley, G. Plant, A. Rostagno, J. Ghiso, B. Frangione: Sporadic and familial cerebral amyloid angiopathies. In: Brain Pathol. 2002 Jul;12(3), S. 343–357. Review. PMID 12146803
- E. E. Smith, S. M. Greenberg: Beta-amyloid, blood vessels, and brain function. In: Stroke. 2009 Jul;40(7), S. 2601–2606. PMID 19443808
- Hereditäre zerebrale Hämorrhagie mit Amyloidose. In: Orphanet (Datenbank für seltene Krankheiten).
- ITM2B-Amyloidose. In: Orphanet (Datenbank für seltene Krankheiten).
- ABeta-Amyloidose Typ Arktis. In: Orphanet (Datenbank für seltene Krankheiten).
- ABeta-Amyloidose Typ Iowa. In: Orphanet (Datenbank für seltene Krankheiten).
- ABetaA21G-Amyloidose. In: Orphanet (Datenbank für seltene Krankheiten).
- ABetaL34V-Amyloidose. In: Orphanet (Datenbank für seltene Krankheiten).
- Abeta-Amyloidose vom holländischen Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- Beta-Amyloidose vom Italienischen Typ. In: Orphanet (Datenbank für seltene Krankheiten).
- K. F. Masuhr, M. Neumann: Neurologie. 6. Auflage. Thieme, Stuttgart 2007, ISBN 978-3-13-135946-9.
- S. M. Greenberg, J. P. Vonsattel: Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. In: Stroke. 1997 Jul;28(7), S. 1418–1422. PMID 9227694
- E. M. Haacke, Z. S. DelProposto, S. Chaturvedi, V. Sehgal, M. Tenzer, J. Neelavalli, D. Kido: Imaging cerebral amyloid angiopathy with susceptibility-weighted imaging. In: AJNR Am J Neuroradiol. 2007 Feb;28(2), S. 316–317. PMID 17297004