Beta-Propeller-Protein-assoziierte Neurodegeneration
Die Beta-Propeller-Protein-assoziierte Neurodegeneration (BPAN[1]) ist eine seltene Krankheit aus dem Spektrum der NBIA-Erkrankungen.[2][3] Mit bis zu 45 % aller Fälle ist sie die häufigste der NBIA-Varianten.[4] BPAN ist durch Entwicklungsverzögerungen und epileptische Anfälle im Kindesalter mit Bewegungsproblemen, einschließlich Dystonie und Parkinsonismus, gekennzeichnet. Sie wird in der Literatur auch als NBIA5 und Static encephalopathy of childhood with neurodegeneration in adulthood (SENDA) bezeichnet. Erste klinische Erwähnungen stammen aus dem Jahr 2012.[5] Mit einer Prävalenz von geschätzt 2–3 pro 1 000 000[6] ist diese Erkrankung sehr selten. In Deutschland sind weniger als 20 Fälle dokumentiert[7] (Stand: September 2018).
| Klassifikation nach ICD-10 | |
|---|---|
| G23.0 | Hallervorden-Spatz-Syndrom |
| ICD-10 online (WHO-Version 2019) | |

Pathogenese
BPAN wird in den meisten Fällen durch eine spontane Mutation (de novo) des WDR45-Gens[8] an der Stelle Xp11.23 des X-Chromosoms verursacht. Bei Mädchen wird der Defekt durch einen dominanten, heterozygoten Erbgang über die Keimbahn hervorgerufen. Bei Jungen findet man entweder eine hemizygote Vererbung durch die Keimbahn oder eine Deletion auf dem WDR45-Gen. Bei einigen Patienten wurden Mosaike festgestellt.[9][10] Die Mutation findet man meist bei Mädchen.[11] Es wird vermutet, dass bei Jungen der Gendefekt so gravierend ist, dass sie das Embryonalstadium meist nicht überleben. In sehr seltenen Fällen, insbesondere wenn die Mutter ein Mosaik der Genveränderung aufweist, kann BPAN von den Eltern vererbt werden.[12] Möglich ist, dass der Anteil der Vererbung über Keimbahnmosaike generell öfter vorkommt als vermutet,[13] so dass zumindest einige solcher Fälle fälschlich als de novo diagnostiziert werden.
Das WDR45-Gen kodiert das Protein WIPI4,[14][15] das eine wichtige Rolle bei der Autophagie in den Nervenzellen des Gehirns spielt.[16][17][18][19] Wegen seiner besonderen Struktur mit sieben flügelartigen Elementen wird es als Beta-Propeller bezeichnet. Durch die Mutation ist die Funktion dieses Proteins beeinträchtigt. Die Schwere der Beeinträchtigung wird offenbar durch den individuellen Gendefekt bestimmt. Studien belegen zudem, dass die Funktion der Mitochondrien sowie des Lysosoms beeinträchtigt ist.[20][21]
Welche Rolle die in den Nervenzellen angesammelten eisenhaltigen Abfallprodukte für das Krankheitsbild spielen, ist nicht vollständig geklärt. Es wird angenommen, dass die durch den Gendefekt beeinträchtigte Autophagie die Symptome dominiert. Neben der oxidativen und daher toxischen Wirkung von freiem Eisen[22] wird vermutet, dass ein Mangel von bioverfügbarem Eisen die Funktion von Zellorganellen wie z. B. die Mitochondrien beeinträchtigt.[23][24][21]
Diagnose
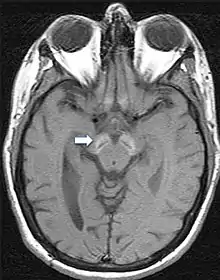

Da die Symptome meist unspezifisch sind, ist eine gesicherte Diagnose nur über bildgebende Verfahren und genetische Sequenzierung möglich. Im fortgeschrittenen Stadium sind mittels Magnetresonanztomographie Veränderungen in der Substantia nigra sowie im Globus pallidus erkennbar, die durch die Anlagerung von Eisen entstehen.[9][25] Frühe Anzeichen vor dem Auftreten von nachweisbaren Eisenablagerungen könnten ein Anschwellen der Substantia nigra sowie eine Hyperintensität in einer T2-gewichteten MRT-Aufnahme des Nucleus dentatus sein.[26] Letztendlich kann die Diagnose jedoch nur durch die Identifizierung der Mutation des WDR45-Gens abgesichert werden. Zur Diagnose der häufig auftretenden Epilepsie sind EEGs angezeigt.
Symptome
Die Symptome[9][27] setzen üblicherweise bereits im frühen Kindesalter oder mit der Geburt ein. Die allgemeine Entwicklung des Kindes ist von Anfang an beeinträchtigt. Nach einer mehrjährigen stabilen Phase erfolgt im frühen Erwachsenenalter eine zunehmende Verschlechterung des kognitiven und allgemeinen Zustands. Die Symptome sind bei den Betroffenen unterschiedlich stark ausgeprägt. Sie beinhalten u. a.:
- globale Entwicklungsverzögerung oder Entwicklungsstörung
- schwere Einschränkung in der Sprachentwicklung
- geistige Behinderung und im späteren Verlauf zunehmender Abbau kognitiver Fähigkeiten bis hin zur Demenz
- Parkinsonismus (Zittern, Bradykinesie, Versteifungen, Instabilitäten)
- Bewegungseinschränkungen (Muskelhypotonie, Muskelhypertonie, Dystonie, Ataxie, Spastik)
- multiple Formen der Epilepsie
- Autismus
- Einschränkung des Sehvermögens
- Schlafstörungen
- Stereotype Bewegungen (ähnlich wie Rett-Syndrom)
- Knirschen (Bruxismus)
- Raynaud-Syndrom
- verzögerte oder unvollständige Myelinisierung im Gehirn
- verfrüht einsetzende Pubertät (Pubertas praecox, Pubarche)
- durch Komplikationen verursachter verfrühter Tod
Die meisten Kinder entwickeln keine Sprache, manche dagegen könne einzelne Wörter oder maximal kurze Sätze formulieren. Das Verständnis von Sprache ist dagegen oft besser entwickelt. Allgemein ist die Kommunikation jedoch schwierig, da es auch kognitive Schwierigkeiten bei der Nutzung von Gesten oder dem Erlernen von Zeichensprache gibt.
Viele Menschen mit BPAN haben wiederkehrende Anfälle (Epilepsie), die im Kindesalter oder in der frühen Kindheit beginnen, sich jedoch allgemein während der Adoleszenz zurückbildet.[6] Sie treten vermehrt beim Übergang zwischen Wachsein und Schlaf auf. Mehrere verschiedene Arten von Anfällen können während dieser Erkrankung auftreten, auch bei der gleichen Person. Häufig treten zuerst Anfälle auf, die durch hohes Fieber ausgelöst werden. Betroffene Personen können auch generalisierte tonisch-klonische Anfälle (auch Grand-Mal-Anfälle genannt) haben. Diese Art von Anfällen betrifft den gesamten Körper und verursacht Muskelstarre, Krämpfe und Bewusstlosigkeit. Andere Anfallsarten, die bei dieser Erkrankung vorkommen können, sind kurze Bewusstseinslücken wie das Auftreten von Abwesenheitsanfällen (auch Petit-Mal-Anfälle genannt), plötzliche Episoden eines schwachen Muskeltonus (atonische Anfälle), unwillkürliche Muskelzuckungen (myoklonische Anfälle) oder ausgeprägtere Bewegungen, die als epileptische Spasmen bezeichnet werden. Einige Patienten haben Anfallsmuster, die denen bei epileptischen Syndromen wie das West-Syndrom oder das Lennox-Gastaut-Syndrom ähneln.
Krankheitsverlauf
Im Gegensatz zu den meisten anderen NBIA-Erkrankungen treten die Symptome bereits mit der Geburt oder in frühester Kindheit auf. Wegen der meist unspezifischen Symptome – insbesondere die Entwicklungsverzögerung – wird die Krankheit zunächst nicht erkannt. Viele Angehörige berichten, dass sowohl Mediziner als auch das persönliche Umfeld oft beschwichtigend reagieren. Mit zunehmendem Alter werden die Einschränkungen jedoch offenbar. Die gängigen Entwicklungsschritte treten später ein oder bleiben aus. Je nach Ausprägung kann dies zu einer schweren Entwicklungsstörung führen, die sich in einer bleibenden Mehrfachbehinderung manifestiert und zu einer Pflegebedürftigkeit führt.
Nach einer Phase von zögerlich einsetzenden Entwicklungsschritten tritt ein Status der Stagnation ein. Etwa im frühen Erwachsenenalter ist eine rapide Verschlechterung des Allgemeinzustands zu erwarten. In einer Studie mit 64 Fällen ergab sich ein Alter von etwa 27 Jahren mit einer erheblichen Schwankung von 13 bis 39 Jahren.[11] Allerdings könnte dieser Zeitkorridor durch eine verzögerte Diagnostik verzerrt sein.[6] Die Dystonien und der kognitive Verfall mit psycho-pathologischen Verhaltensänderungen nehmen bis hin zur Demenz zu. Durch Komplikationen kann ein verfrühter Tod eintreten.
Jedoch zeigt sich, dass es keine strikt einheitliche Entwicklung von BPAN gibt, so dass der exakte Krankheitsverlauf bisher nicht prognostizierbar ist. Die verschiedenen Ausprägungen können sehr unterschiedlich ausfallen und sind derzeit noch in der Erforschung.
Therapie

Eine spezielle Therapie oder gar Heilung von BPAN existiert derzeit nicht. Die Behandlungen beschränken sich auf die verschiedenen Symptome.[6] Sie schließen Medikamente und therapeutische Anwendungen ein.
Epilepsie
Die Formen der Epilepsie können sehr unterschiedlich sein. Auch kann ein Patient unter verschiedenen Varianten der Epilepsie leiden. Daher ist es wichtig, die oft komplexe Anfallstätigkeit gut zu diagnostizieren und entsprechend zu behandeln. Diese beinhaltet in erster Linie Medikamente. Viele Betroffene berichten von einer positiven Wirkung einer ketogenen Diät, die ebenfalls von Medizinern zur Verbesserung des Zustands empfohlen wird[28][29][30]. Da sich die Anfallstätigkeit häufig mit zunehmendem Alter abschwächt,[9] ist eine regelmäßige Kontrolle sowie eine entsprechende Anpassung angezeigt.
Störung der Sprachentwicklung
Die Entwicklung von gesprochener Sprache lässt sich offenbar kaum beeinflussen. Dennoch profitieren die Betroffenen von Logopädie, die die Atmung und das Kauen bei der Nahrungsaufnahme schult. Unterstützte Kommunikation kann bei der Überwindung von Verständnisbarrieren helfen. Auch können technische Hilfsmittel wie Computer die Entwicklung der Kommunikation begünstigen. Allerdings werden diese Ansätze oft durch kognitive Beeinträchtigungen erschwert, da dem Patienten womöglich zunächst ein Verständnis von Ursache und Wirkung vermittelt werden muss. Hier können Geräte wie ein Step-by-Step nützlich sein. Ergotherapie fördert die kognitive Entwicklung ebenfalls.
Bewegungseinschränkungen
Physiotherapie sollte so früh wie möglich in Anspruch genommen werden. Sie kann die Beweglichkeit der Muskeln und Gelenke verbessern und fördern. Je nach Einschränkung können Hilfsmittel wie Geh- und Stehtrainer sowie Orthesen den Zustand verbessern. Bei anhaltenden Verkrampfungen wird manchmal Botox in den Muskel gespritzt, was zu einer zeitweisen Erleichterung des Krampfzustands führt.[31] Der während der Progression zunehmende Parkinsonismus kann gut mit dopaminerger Medikation wie Levodopa behandelt werden.[32]
Verhaltensänderungen
Beim Übergang in das Erwachsenenalter ist eine Verschlechterung des Allgemeinzustands zu erwarten. Neben der schon erwähnten Zunahme von Parkinsonismus und Dystonien tritt oft eine zunächst subtile Verhaltensänderung ein, die von Apathie und Aggression bis hin zur Demenz reichen. Hier sind psychotherapeutische Maßnahmen angezeigt.[6]
Ernährung, Diäten und Nahrungsergänzungsmittel
Es existieren keine Hinweise auf einen positiven Einfluss von speziellen Diäten oder der Verwendung von Nahrungsergänzungsmitteln. Von einer eisenarmen Ernährung wird abgeraten, da sie keinen Einfluss auf den Eisenhaushalt im Gehirn hat. Die Evidenzen für eine positive Wirkung von Cannabidiol auf Stimmungsschwankungen, Autismussymptome, Schlafstörungen und Verhaltensstörungen sind gering. Solche Stoffe sollten nur in Absprache mit Fachleuten verabreicht werden. Bei Schluckstörungen kann der Einsatz einer Magensonde oder einer PEG-Sonde notwendig werden.[6]
Forschungs- und Therapieansätze
Medizinische Studien haben gezeigt, dass der Versuch einer Reduktion von Eisen in den Hirnzellen keinen signifikanten Einfluss auf die Gesundheit der Patienten hat.[33][34] In einer Fallstudie führte die Gabe des Chelatbildners Deferipron sogar zu einer deutlichen Verschlechterung der Symptomatik, sodass die Behandlung abgebrochen werden musste.[35]
Wegen der Verbindung zum Prozess der Autophagie, der durch die Mutation des WDR45-Gens gehemmt wird, konzentrieren sich einige vorklinische Studien, auch in verwandten Erkrankungen wie Parkinson und Alzheimer, auf eine Verstärkung dieses Mechanismus. Ein Angriffspunkt ist hierbei das Protein mTOR, das selbst die Autophagie dämpft[36]. Substanzen wie das Immunsuppressivum Rapamycin hemmen mTOR[37], wodurch die Autophagie angeregt wird. Rapamycin wird derzeit in mehreren zellulären und Tierstudien auf seine Eignung als Therapeutikum untersucht.[38][39] In einem BPAN-Mausmodell konnte Rapamycin teilweise die Autophagie restaurieren.[40]
Während mTOR an einer frühen Stelle des Autophagieprozesses einsetzt, gibt es Anzeichen dafür, dass Mutationen von WDR45 und WDR45b die Fusion der Autophagosome mit den Lysosomen stören und somit den Abbau der Zellprodukte unterbinden.[41] Eine Aktivierung dieser Fusion könnte ein weiterer Ansatz für eine kausale Therapie sein.
Ein weiterer Zusammenhang zwischen Parkinson und Neurodegeneration mit Eisenablagerung im Gehirn im Allgemeinen und BPAN im Besonderen scheint im Dopamin-Metabolismus und Alpha-Synuclein zu bestehen.[42]
Lebenserwartung und Palliativversorgung
Die Lebenserwartung hängt sehr stark vom individuellen Verlauf der Erkrankung ab. Mit der Zunahme von Dystonien bei erwachsenen Patienten in einem Altersbereich zwischen ungefähr 30 und 50 Jahren erhöht sich auch das Sterberisiko.[43] Todesfälle gehen meist auf Komplikationen im Zusammenhang mit Parkinsonismus und Aspiration zurück.[2] Insbesondere bei fortschreitender Verschlechterung des Allgemeinzustands mit schmerzhaften Dystonien sollte eine Versorgung in Erwägung gezogen werden, die sich auf eine Verbesserung der Lebensqualität konzentriert.[6]
Interessensverbände und Selbsthilfegruppen
- Hoffnungsbaum e. V.
- Millys Mission
- BPAN Warriors
- NBIA Disorders Association
- NBIA cure
- NBIA Alliance
- EURORDIS
- ACHSE e. V.
Die deutsche Patientenorganisation Hoffnungsbaum e. V. hat gemeinsam mit ACHSE e. V., der Dachorganisation für seltene Erkrankungen, im Jahre 2017 eine patientenorientierte Beschreibung zu NBIA veröffentlicht, die noch einige weitere Details und Informationen enthält.
Weblinks
- Patientenorientierte Krankheitsbeschreibung aus dem ACHSE Netzwerk: NBIA–Erkrankungen. (PDF) ACHSE e. V, abgerufen am 25. Januar 2019.
- Sonderforschungsbereich 1177: Molecular and Functional Characterization of Selective Autophagy. Projekt E03: Molecular functions of WDR45/WlPI4 in ferritinophagy and neurodegeneration. Abgerufen am 19. Oktober 2021.
Einzelnachweise
- Beta-Propeller-Protein-assoziierte Neurodegeneration. In: Online Mendelian Inheritance in Man. (englisch)
- Susan J. Hayflick, Michael C. Kruer, Allison Gregory, Tobias B. Haack, Manju A. Kurian: Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. In: Brain. Band 136, Nr. 6, 1. Juni 2013, ISSN 0006-8950, S. 1708–1717, doi:10.1093/brain/awt095, PMID 23687123, PMC 3673459 (freier Volltext) – (oup.com [abgerufen am 28. April 2018]).
- Einführung und Klinische Symptomatik. In: NBIA – Krankheitsbilder. Friedrich-Baur-Institut, Klinikum der Universität München, abgerufen am 28. April 2018.
- Allison Gregory, Susan Hayflick: Neurodegeneration with Brain Iron Accumulation Disorders Overview. In: GeneReviews®. University of Washington, Seattle, Seattle (WA) 2013, PMID 23447832 (englisch, nih.gov [abgerufen am 7. November 2019]).
- Tobias B. Haack, Penelope Hogarth, Michael C. Kruer, Allison Gregory, Thomas Wieland: Exome Sequencing Reveals De Novo WDR45 Mutations Causing a Phenotypically Distinct, X-Linked Dominant Form of NBIA. In: The American Journal of Human Genetics. Band 91, Nr. 6, Dezember 2012, ISSN 0002-9297, S. 1144–1149, doi:10.1016/j.ajhg.2012.10.019, PMID 23176820, PMC 3516593 (freier Volltext) – (elsevier.com [abgerufen am 28. April 2018]).
- Jenny L Wilson, Allison Gregory, Manju A Kurian, Ittai Bushlin, Fanny Mochel: Consensus clinical management guideline for beta-propeller protein-associated neurodegeneration. In: Bernard Dan (Hrsg.): Developmental Medicine & Child Neurology. Mac Keith Press, 4. August 2021, ISSN 0012-1622, doi:10.1111/dmcn.14980, PMID 34347296 (englisch).
- B. Büchner, Friedrich-Baur-Institut, Klinikum der Ludwig-Maximilians-Universität München, mündl. Kommunikation, 21. September 2018.
- WDR45 gene. In: Genetics Home Reference. U. S. National Library of Medicine, abgerufen am 13. Mai 2018 (englisch).
- Allison Gregory, Manju A. Kurian, Tobias Haack, Susan J. Hayflick, Penelope Hogarth: Beta-Propeller Protein-Associated Neurodegeneration. In: Margaret P. Adam, Holly H. Ardinger, Roberta A. Pagon, Stephanie E. Wallace (Hrsg.): GeneReviews® [Internet]. University of Washington, 16. Februar 2017, ISSN 2372-0697, PMID 28211668 (nih.gov).
- Beta-propeller protein-associated neurodegeneration. In: Genetics Home Reference. NIH, U.S. National Library of Medicine, abgerufen am 27. April 2018.
- Kjersti Eline Stige, Ivar Otto Gjerde, Gunnar Houge, Per Morten Knappskog, Charalampos Tzoulis: Beta-propeller protein-associated neurodegeneration: a case report and review of the literature. In: Clinical Case Reports. Band 6, Nr. 2, 4. Januar 2018, ISSN 2050-0904, S. 353–362, doi:10.1002/ccr3.1358, PMID 29445477, PMC 5799652 (freier Volltext).
- Yuri A Zarate, Julie R Jones, Melanie A Jones, Francisca Millan, Jane Juusola: Lessons from a pair of siblings with BPAN. In: European Journal of Human Genetics. Band 24, Nr. 7, 18. November 2015, ISSN 1018-4813, S. 1080–1083, doi:10.1038/ejhg.2015.242, PMID 26577041, PMC 5070893 (freier Volltext).
- Ian M. Campbell, Bo Yuan, Caroline Robberecht, Rolph Pfundt, Przemyslaw Szafranski: Parental Somatic Mosaicism Is Underrecognized and Influences Recurrence Risk of Genomic Disorders. In: American Journal of Human Genetics. Band 95, Nr. 2, 7. August 2014, ISSN 0002-9297, S. 173–182, doi:10.1016/j.ajhg.2014.07.003, PMID 25087610, PMC 4129404 (freier Volltext).
- Tassula Proikas-Cezanne, Scott Waddell, Anja Gaugel, Tancred Frickey, Andrei Lupas: WIPI-1α (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. In: Oncogene. Band 23, Nr. 58, Dezember 2004, ISSN 0950-9232, S. 9314–9325, doi:10.1038/sj.onc.1208331.
- T. Proikas-Cezanne, Z. Takacs, P. Donnes, O. Kohlbacher: WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. In: Journal of Cell Science. Band 128, Nr. 2, 15. Januar 2015, ISSN 0021-9533, S. 207–217, doi:10.1242/jcs.146258.
- Daniela Bakula, Amelie J. Müller, Theresia Zuleger, Zsuzsanna Takacs, Mirita Franz-Wachtel, Ann-Katrin Thost, Daniel Brigger, Mario P. Tschan, Tancred Frickey, Horst Robenek, Boris Macek & Tassula Proikas-Cezanne: WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. In: Nature Communications. Band 8, 31. Mai 2017, ISSN 2041-1723, S. 15637, doi:10.1038/ncomms15637, PMID 28561066, PMC 5460038 (freier Volltext).
- Saikat Chowdhury, Chinatsu Otomo, Alexander Leitner, Kazuto Ohashi, Ruedi Aebersold, Gabriel C. Lander, Takanori Otomoa: Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. In: Proceedings of the National Academy of Sciences of the United States of America. Band 115, Nr. 42, 5. September 2018, ISSN 1091-6490, S. E9792–E9801, doi:10.1073/pnas.1811874115, PMID 30185561, PMC 6196511 (freier Volltext).
- Takanori Otomo, Saikat Chowdhury, Gabriel C. Lander: The Rod-Shaped ATG2A-WIPI4 Complex Tethers Membranes In Vitro. In: Contact. Band 1, 21. Dezember 2018, ISSN 2515-2564, doi:10.1177/2515256418819936, PMID 30766969, PMC 6372110 (freier Volltext).
- Shintaro Maeda, Chinatsu Otomo, Takanori Otomo: The autophagic membrane tether ATG2A transfers lipids between membranes. In: eLife. Band 8, 4. Juli 2019, ISSN 2050-084X, doi:10.7554/eLife.45777, PMID 31271352, PMC 6625793 (freier Volltext).
- Philip Seibler, Lena F Burbulla, Marija Dulovic, Simone Zittel, Johanne Heine: Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. In: Brain. Band 141, Nr. 10, 30. August 2018, ISSN 0006-8950, S. 3052–3064, doi:10.1093/brain/awy230 (oup.com [abgerufen am 28. September 2018]).
- Angelika Klucken, Lena Burbulla: Lena Burbulla entdeckt neue mögliche BPAN-Krankheitsmechanismen. Hoffnungsbaum e. V., 2. August 2021, abgerufen am 8. August 2021.
- Geon Ha Kim, Jieun E. Kim, Sandy Jeong Rhie, Sujung Yoon: The Role of Oxidative Stress in Neurodegenerative Diseases. In: C. Justin Lee, Jong Eun Lee, Byung-Ok Choi (Hrsg.): Experimental Neurobiology. Band 24, Nr. 4, 12. Oktober 2015, ISSN 1226-2560, S. 325, doi:10.5607/en.2015.24.4.325, PMID 26713080, PMC 4688332 (freier Volltext).
- Finanziato un progetto sulla BPAN: Lena Burbulla studierà il legame tra ferro e neurodegenerazione. In: Associazione Italiana Sindromi Neurodegenerative da Accumulo di Ferro. 17. August 2019, abgerufen am 22. August 2019 (italienisch).
- Finanzierung BPAN-Projekt zum Zusammenhang zwischen Eisen und Neurodegeneration. Hoffnungsbaum e. V., 21. August 2019, abgerufen am 22. August 2019.
- Yuta Ichinose, Michiaki Miwa, Akiko Onohara, Kimiko Obi, Kazumasa Shindo: Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). In: Neurology: Clinical Practice. Band 4, Nr. 2, 1. April 2014, ISSN 2163-0402, S. 175–177, doi:10.1212/01.CPJ.0000437694.17888.9b, PMID 24790802, PMC 4001181 (freier Volltext) – (neurology.org [abgerufen am 28. April 2018]).
- Camilla Russo, Anna Ardissone, Elena Freri, Serena Gasperini, Marco Moscatelli, Giovanna Zorzi, Celeste Panteghini, Barbara Castellotti, Barbara Garavaglia, Nardo Nardocci, Luisa Chiapparini: Substantia Nigra Swelling and Dentate Nucleus T2 Hyperintensity May Be Early Magnetic Resonance Imaging Signs of β-Propeller Protein-Associated Neurodegeneration: Early MRI Features in 4 Cases of BPAN. In: Kailash Bhatia, Marcelo Merello, International Parkinson and Movement Disorder Society (Hrsg.): Movement Disorders - Clinical Practice. Band 6, Nr. 1. John Wiley & Sons, Inc., 10. Oktober 2018, S. 51–56, doi:10.1002/mdc3.12693, PMID 30746416, PMC 6335537 (freier Volltext).
- BPAN. In: NBIA Disorders. NBIA cure, abgerufen am 28. April 2018 (englisch).
- Ketogene Diät bei resistenter Epilepsie wirksam. In: aerzteblatt.de. Deutscher Ärzteverlag GmbH, 8. Mai 2008, abgerufen am 9. August 2021.
- Elizabeth G Neal, Hannah Chaffe, Ruby H Schwartz, Margaret S Lawson, Nicole Edwards: The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. In: The Lancet Neurology. Band 7, Nr. 6, Juni 2008, ISSN 1474-4422, S. 500–506, doi:10.1016/s1474-4422(08)70092-9.
- Eunjoo Lee, Hoon-Chul Kang, Heung Dong Kim: Ketogenic Diet for Children with Epilepsy: A Practical Meal Plan in a Hospital. In: Clinical Nutrition Research. Band 5, Nr. 1, 2016, ISSN 2287-3732, S. 60, doi:10.7762/cnr.2016.5.1.60, PMID 26839878, PMC 4731863 (freier Volltext).
- Botulinumtoxin (Injektionstherapie). Deutsche Dystonie Gesellschaft e. V., abgerufen am 27. Januar 2019.
- Penelope Hogarth: Neurodegeneration with brain iron accumulation: diagnosis and management. In: Journal of Movement Disorders. Band 8, Nr. 1, Januar 2015, ISSN 2005-940X, S. 1–13, doi:10.14802/jmd.14034, PMID 25614780, PMC 4298713 (freier Volltext).
- Petr Dusek, Susanne A. Schneider, Jan Aaseth: Iron chelation in the treatment of neurodegenerative diseases. In: Dirk Schaumlöffel (Hrsg.): Journal of Trace Elements in Medicine and Biology. Band 38. Elsevier, Dezember 2016, ISSN 0946-672X, S. 81–92, doi:10.1016/j.jtemb.2016.03.010 (sciencedirect.com [abgerufen am 25. September 2018]).
- Shen-Yang Lim, Ai Huey Tan, Azlina Ahmad-Annuar, Susanne A. Schneider, Ping Chong Bee: A Patient with Beta-Propeller Protein-Associated Neurodegeneration: Treatment with Iron Chelation Therapy. In: Journal of Movement Disorders. Band 11, Nr. 2, Mai 2018, ISSN 2005-940X, S. 89–92, doi:10.14802/jmd.17082, PMID 29860786, PMC 5990906 (freier Volltext) – (englisch).
- Mattia Fonderico, Michele Laudisi, Nico Golfrè Andreasi, Stefania Bigoni, Costanza Lamperti et al.: Patient Affected by Beta-Propeller Protein-Associated Neurodegeneration: A Therapeutic Attempt with Iron Chelation Therapy. In: Frontiers in Neurology. Band 8, 2017, ISSN 1664-2295, S. 385, doi:10.3389/fneur.2017.00385, PMID 28878728, PMC 5573443 (freier Volltext) – (englisch).
- Joungmok Kim, Mondira Kundu, Benoit Viollet, Kun-Liang Guan: AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. In: Nature Cell Biology. Band 13, Nr. 2, Februar 2011, ISSN 1465-7392, S. 132–141, doi:10.1038/ncb2152, PMID 21258367, PMC 3987946 (freier Volltext) – (englisch, nature.com [abgerufen am 19. Oktober 2021]).
- Dudley W. Lamming: Inhibition of the Mechanistic Target of Rapamycin (mTOR)–Rapamycin and Beyond. In: Cold Spring Harbor Perspectives in Medicine. Band 6, Nr. 5, Mai 2016, ISSN 2157-1422, S. a025924, doi:10.1101/cshperspect.a025924, PMID 27048303, PMC 4852795 (freier Volltext) – (englisch, cshlp.org [abgerufen am 19. Oktober 2021]).
- Jordi Bové, Marta Martínez-Vicente, Miquel Vila: Fighting neurodegeneration with rapamycin: mechanistic insights. In: Nature Reviews Neuroscience. Band 12, Nr. 8, August 2011, ISSN 1471-003X, S. 437–452, doi:10.1038/nrn3068 (englisch, nature.com [abgerufen am 19. Oktober 2021]).
- resTORbio Announces Interim Results for Phase 1b/2a Trial of RTB101 in Patients with Parkinson’s Disease and Provides Corporate Update. In: Globenewswire. Adicet Bio, 19. Februar 2020, abgerufen am 19. Oktober 2021 (englisch).
- Huida Wan, Qi Wang, Xiuting Chen, Qiufang Zeng, Yanjiao Shao: WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. In: Autophagy. Band 16, Nr. 3, 3. März 2020, ISSN 1554-8627, S. 531–547, doi:10.1080/15548627.2019.1630224, PMID 31204559, PMC 6999610 (freier Volltext) – (englisch, tandfonline.com [abgerufen am 19. Oktober 2021]).
- Cuicui Ji, Hongyu Zhao, Di Chen, Hong Zhang, Yan G. Zhao: β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. In: Current Biology. Band 31, Nr. 8, April 2021, S. 1666–1677.e6, doi:10.1016/j.cub.2021.01.081 (englisch, elsevier.com [abgerufen am 19. Oktober 2021]).
- Rachel M. Wise, Annika Wagener, Urban M. Fietzek, Thomas Klopstock, Eugene V. Mosharov, Fabio A. Zucca, David Sulzer, Luigi Zecca, Lena F. Burbulla: Interactions of dopamine, iron, and alpha-synuclein linked to dopaminergic neuron vulnerability in Parkinson's disease and Neurodegeneration with Brain Iron Accumulation disorders. In: Dr. Erwan Bezard et al. (Hrsg.): Neurobiology of Disease. Band 175. Elsevier, Dezember 2022, ISSN 0969-9961, S. 105920, doi:10.1016/j.nbd.2022.105920, PMID 36351559 (englisch, elsevier.com).
- Afshin Saffari, Julian Schröter, Sven F. Garbade, Julian E. Alecu, Darius Ebrahimi-Fakhari, Georg F. Hoffmann, Stefan Kölker, Markus Ries, Steffen Syrbe: Quantitative retrospective natural history modeling of WDR45 -related developmental and epileptic encephalopathy – a systematic cross-sectional analysis of 160 published cases. In: Autophagy. Taylor & Francis Online, 24. November 2021, ISSN 1554-8627, S. 1–13, doi:10.1080/15548627.2021.1990671, PMID 34818117 (tandfonline.com [abgerufen am 9. Februar 2022]).