Paraffinoxidation
Die Paraffinoxidation ist ein historisches chemisch-technisches Verfahren zur Herstellung synthetischer Fettsäuren, welche die chemische Industrie sowohl zu Konsumgütern wie Seifen und Speisefetten als auch zu Schmierfetten für technische Anwendungen verarbeitete. Als weitere Produkte fielen ein breites Spektrum von Carbonsäuren und Oxidationsprodukte wie Alkohole, Aldehyde, Ester oder Ketone an. Rohstoffbasis war kohlestämmiges Paraffingatsch, ein gesättigtes, höhermolekulares Kohlenwasserstoffgemisch und Nebenprodukt der Fischer-Tropsch-Synthese. Die Oxidation der Paraffine erfolgte im flüssigen Zustand durch molekularen Luftsauerstoff unter Spaltung der Kohlenstoffkette in Anwesenheit von Permanganaten, bei Temperaturen im Bereich von etwa 100 bis 120 °C und unter Normaldruck.

Die Produkte der Paraffinoxidation standen in Konkurrenz zu natürlich vorkommenden Fettsäuren. Verknappungen durch Konflikte oder Engpässe in der Lebensmittelversorgung verstärkten das Interesse an synthetischen Fettsäureerzeugnissen. In Deutschland erlangte das Verfahren ab Mitte der 1930er Jahre im Rahmen der Autarkiebestrebungen des Deutschen Reichs kommerzielle Bedeutung und wurde bis in die ersten Jahre nach dem Zweiten Weltkrieg im großindustriellen Maßstab durchgeführt. Mit diesem Verfahren wurde der Rohstoff für den technischen Fett- und Waschmittelsektor hergestellt; die dafür zuvor benötigten nativen Fette standen damit dem Ernährungssektor zur Verfügung. Weiterhin galt die durch die Paraffinoxidation erstmals ermöglichte großtechnische Erzeugung von künstlichen, für die menschliche Ernährung geeigneten Fetten („Butter“) aus Kohle in der damaligen Zeit als Sensation.
Durch die hohe Verfügbarkeit von preiswerten nativen Fetten sowie die Konkurrenz durch erdölstämmige Fettalkohole verlor das Verfahren Anfang der 1950er Jahre in der westlichen Welt an Bedeutung. Verfahrensvarianten wie die Bashkirov-Oxidation, bei der Alkane ohne Kettenspaltung in Gegenwart von Borsäure zu sekundären Alkoholen oxidiert werden, wendet die chemische Industrie aber bis heute für spezielle Synthesen an, beispielsweise für die Produktion von Cyclododecanol.
Geschichte

Frühe Arbeiten
Paraffine sind ein Gemisch aus Alkanen, gesättigten Kohlenwasserstoffen mit der allgemeinen Summenformel CnH2n+2. Ihr Name leitet sich von lateinisch parum affinis, wenig beteiligt, ab. Paraffine galten als wenig reaktionsfreudig; mit Reduktionsmitteln wie metallischem Natrium beispielsweise reagieren Paraffine bei Umgebungstemperatur nicht.
Im Jahr 1854, nur 19 Jahre nach der Entdeckung der Paraffine durch Karl von Reichenbach,[1] wies Gotthard Hofstädter darauf hin, dass die Paraffine nicht so reaktionsträge sind, wie ihr Name suggeriert. Er berichtete erstmals über Versuche zur Oxidation von Paraffinen durch Kochen mit Salpetersäure. Als Hauptprodukt erhielt er dabei Bernsteinsäure, eine Dicarbonsäure, daneben niedermolekulare, wasserlösliche Monocarbonsäuren.[2] Auch andere starke Oxidationsmittel, wie die von Eduard Meusel verwendete Chromsäure, führten zur Bildung von Carbonsäuren.[3] 1874 beschrieb A. G. Pouchet die Paraffinoxidation mittels eines Gemischs aus rauchender Salpeter- und Schwefelsäure bei Temperaturen unterhalb von 110 °C. Als Produkte fielen erneut eine Reihe von niedermolekularen, wasserlöslichen Carbonsäuren an. Daneben identifizierte Pouchet erstmals eine neue höhermolekulare Carbonsäure, eine Fettsäure, die er „Paraffinsäure“ nannte.[4]
Oxidation mit Luft
Pompejus Bolley erkannte 1868, dass Paraffine bei einer Temperatur von 150 °C den Luftsauerstoff absorbierten,[5] Carl Engler stellte 1879 bei ähnlichen Untersuchungen die Bildung von wasserlöslichen Fettsäuren fest.[6] Die praktische Bedeutung ihrer Beobachtungen erkannten die Forscher jedoch nicht. Erst einige Jahre später identifizierte Eugen Schaal das kommerzielle Potential dieser Reaktion. Er reichte 1884 das erste Patent für die Umwandlung von Petroleum und ähnlichen Kohlenwasserstoffen in Carbonsäuren durch Oxidation mit Luft ein. Als Katalysator nutzte Schaal unter anderem auf Kieselgur adsorbierte Chlorate, Permanganate oder Nitrate.[7] Die Verwendung von preiswertem Luftsauerstoff stellte einen wesentlichen Fortschritt auf dem Wege zur technischen Umsetzung des Verfahrens dar. Da bis zum Ausbruch des Ersten Weltkriegs Fettsäuren aus natürlichen Quellen ausreichend zur Verfügung standen, rechnete sich eine Kommerzialisierung dieser Verfahren zunächst nicht.
Erst als während des Weltkrieges Fette und damit Fettsäuren zur Mangelware wurden, verstärkte die chemische Industrie die Forschungsanstrengungen, um zumindest den Rohstoff für die Seifenproduktion auf Grundlage von Petroleum oder den bei der Schwelung von Braunkohle anfallenden Teer zu gewinnen. Versuche, die damals bereits bekannten Verfahren in die industrielle Praxis zu überführen, gelangen nur bedingt.[8] Das erste technische Verfahren während des Krieges betrieb die Firma Fanto in Pardubice und nutzte ein Quecksilbersalz als Katalysator. Die erzeugten Fettsäuren waren jedoch von geringer Qualität, die Umsetzung der Paraffine dauerte Tage. Eine Verbesserung von Ausbeute und Qualität gelang durch das systematische Studium des Temperatureinflusses sowie der Luftmenge auf die Qualität des Oxidationsproduktes durch Adolf Grün bei der Schicht AG.[9] Die technisch von der Schicht AG hergestellten Fettsäuren ließen sich gut verseifen, die Seifen lieferten befriedigende Waschwirkung.[10]
Nach dem Ersten Weltkrieg in den 1920er Jahren setzten größere Chemieunternehmen wie die IG Farben sowie die Firmen Henkel und Hubbe & Fahrenholz, damals eine der größten und bedeutendsten Ölmühlen in Deutschland, die Forschung auf dem Gebiet der Paraffinoxidation fort. Eine Produktionsstätte der IG Farben in Ludwigshafen stellte ab 1928 synthetische Fettsäuren im kleineren Maßstab her, ein Gemeinschaftsunternehmen der IG Farben und Standard Oil of New Jersey in Baton Rouge produzierte 1931 etwa 3 Tonnen Fettsäuren pro Tag auf Basis von petroleumstämmigen Paraffinen.[10] Die bei dem Verfahren gewonnenen Fettsäuren führten jedoch in der Weiterverarbeitung zu Seifen minderer Qualität mit Geruchsproblemen. Da außerdem die Rohstoffbasis Paraffine knapp und damit teuer war, boten die Verfahren im Vergleich mit den Kosten für die Beschaffung nativer Fettsäuren zu diesem Zeitpunkt wiederum keinen ökonomischen Vorteil.[10]
Deutsche Fettsäure-Werke
Der Kolloidchemiker Arthur Imhausen, Mitinhaber der Märkischen Seifenindustrie, nahm in den 1930er Jahren die Forschungen von Eugen Schaal wieder auf. Mit seinem Mitarbeiter Werner Prosch entwickelte er das Imhausen-Prosch-Verfahren zur Paraffinoxidation. Das Verfahren verwendete Luft als Oxidationsmittel und Kaliumpermanganat als Initiator. Zunächst verwandte Imhausen Paraffine aus der Hydrierung von Braunkohleschwelprodukten. Der Kommissar für Wirtschaftsfragen Wilhelm Keppler, der sowohl für die Durchführung des Vierjahresplans mit dem Schwerpunkt „Industrielle Fette und Öle“ als auch für die Förderung der Benzinerzeugung zuständig war, bot Imhausen die Verwendung des bei der Fischer-Tropsch-Synthese anfallenden Paraffingatschs als Rohstoff an.[11] Beim Paraffingatsch handelte es sich um eine Mischung von Alkanen ohne wesentliche Verunreinigungen, das bis dahin als störendes Nebenprodukt der Fischer-Tropsch-Synthese galt. Es erwies sich als idealer Rohstoff für die Paraffinoxidation. Durch die Verwertbarkeit aller Kohlesorten in der Fischer-Tropsch-Synthese bestand eine breite Rohstoffbasis für die Gatschherstellung.[12] Weiterhin übernahm das Reich sowohl Preisgarantien für den gelieferten Rohstoff als auch für die produzierten Fettsäuren.[12]
Bezogen auf den Paraffingatsch erzielte Imhausen eine Ausbeute bei den Zielprodukten, den Hauptlauffettsäuren mit einer Kettenlänge von 11 bis 20 Kohlenstoffatomen von etwa 50 %. Für eine großtechnische Umsetzung fehlte Imhausen das notwendige Kapital, außerdem gab es technische Probleme bei der Aufarbeitung des Rohfettsäuregemischs. Da Henkel ebenfalls Interesse an Fettsäuren hatte und über ausreichende finanzielle Mittel verfügte, gründeten Imhausen und Henkel am 21. Februar 1936 die Deutsche Fettsäure-Werke GmbH in Witten als Gemeinschaftsunternehmen zur Herstellung und Vermarktung von Fettsäuren nach dem Imhausen-Prosch-Verfahren.[11] Die Gesellschafter beriefen Arthur Imhausen und Erich Grünthal zu Geschäftsführern, Vorsitzender des Aufsichtsrates wurde der Persil-Erfinder Hugo Henkel.[11]
Da auch die IG Farben ein Verfahren zur Paraffinoxidation entwickelt hatte, erfolgte auf Vermittlung von Keppler eine Kooperation zwischen den Deutschen Fettsäure-Werken und der IG Farben zwecks Optimierung der Produktionsverfahren.[11] Das IG Farben-Verfahren bot Vorteile bei der Weiterverarbeitung der Rohfettsäuren. Ab etwa Mai 1938 überwand Imhausen die technischen Schwierigkeiten der Rohfettsäureverarbeitung mit Hilfe des Verfahrens der IG Farben und erhielt ein qualitativ hochwertiges Produkt. Da die Fischer-Tropsch-Kraftstoffe eine niedrige Oktanzahl aufwiesen, baute die nationalsozialistische Wirtschaftsführung stattdessen mehr Hydrieranlagen nach dem Bergius-Pier-Verfahren, die ein höherwertiges Motorenbenzin lieferten; Paraffingatsch wurde schwer erhältlich. Erst ab 1940 steigerten die Fischer-Tropsch-Anlagen ihren Ausstoß erheblich, was zu einer guten Versorgung mit Gatsch führte. Auf Veranlassung von Imhausen ließ Keppler die Fischer-Tropsch-Anlagen auf das Mitteldruckverfahren umstellen, das eine höhere Ausbeute an Paraffingatsch lieferte.[12] Die Werke Ludwigshafen-Oppau und Heydebreck O.S. der IG Farben produzierten je 20.000, das Werk Witten 40.000 Tonnen Fettsäuren pro Jahr.[13] Während das Werk Witten fast nur Fischer-Tropsch-Gatsch verarbeitete, verbrauchte das Werk Oppau zu etwa 80 % braunkohlestämmige TTH-Paraffine, 10 % Fischer-Tropsch-Gatsch und 10 % Nerag-Gatsch, welches aus der Stockpunkterniedrigung von Spindelöl stammte.[14]
Butter aus Kohle

Vor dem Beginn des Zweiten Weltkriegs deckte die Inlandserzeugung in Deutschland weniger als die Hälfte des Nahrungsfettbedarfs von circa 1,6 Millionen Tonnen, auf dem technischen Sektor mit einem Bedarf von circa 400.000 Tonnen nur etwa 13 %.[15] Den Restbedarf deckte Deutschland durch den Import von Ölen und Fetten von Ölpflanzen aus Ostasien und Südamerika sowie durch arktische Walöle.[8] Die politische Absicht, die Abhängigkeit des Deutschen Reiches vom Import technischer Fette und Nahrungsfette zu beenden, führte bald zur sogenannten Fettlücke. Ab August 1939, vier Tage vor Kriegsbeginn, rationierten die Nationalsozialisten den Bezug von Fetten und steuerten deren Ausgabe über eine Reichsfettkarte.[16]
Arthur Imhausen, der jüdischer Abstammung war, gelang es, aus den reinen Fettsäuren der Paraffinoxidation durch Veresterung mit Glycerin das erste synthetische Speisefett der Welt herzustellen und damit die Autarkiebestrebungen des Dritten Reichs zu unterstützen. Auf Vorschlag von Hermann Göring erkannte Adolf Hitler daraufhin Imhausens Familie als Arier an.[17] Ab 1941 stellten die Deutschen Fettwerke Witten in einer Großanlage monatlich 250 Tonnen künstliches Speisefett her.[18] Auf Wilhelm Kepplers Rat hin teilten die Nationalsozialisten das Kunstfett versuchsweise etwa drei Jahre lang an Häftlinge des Konzentrationslagers Sachsenhausen und in den Lagern des Reichsarbeitsdienstes Trebbin und Ruhlsdorf aus.[19][15] Mediziner der Physiologischen Abteilung des Reichsgesundheitsamts führten daneben Fütterungsversuche an Tieren durch.[20] Als bedenklich galt das Auftreten von ungeradzahligen Fettsäuren, die in nativen Fetten nur selten vorkommen. Negative physiologischen Auswirkungen konnten jedoch nicht nachgewiesen werden.[21] Anteile an ethylverzweigten Carbonsäuren im Fett, sogenannte Isofettsäuren, führten dagegen zur gesundheitlich bedenklichen Dicarbonsäurebildung, die sich schon bei geringer Gabe im Urin nachweisen ließ.[21] Nach dreijähriger Prüfung gab das Reichsgesundheitsamt 1939, wenige Tage nach dem Überfall der deutschen Wehrmacht auf Polen, unter politischem Druck die Kunstbutter zum Verzehr frei.[15] Das Fett diente als Zusatz zu Schwerarbeiterrationen, zum Kantinenessen in Spitälern und ergänzte die Rationen der Insassen von Arbeits- und Konzentrationslagern sowie von sowjetischen Kriegsgefangenen.[15] Während des Afrikafeldzugs und auf den deutschen Unterseebooten versorgte das Militär die Soldaten mit Speisefett aus der Wittener Produktion. Das Fett wurde durch den hohen Anteil an gesättigten Fettsäuren kaum ranzig und war daher lange haltbar. Geschmacklich kam es der Butter nahe und es hatte denselben Brennwert.[22] Die Hoffnungen Imhausens auf eine Produktionsmenge jenseits von 100.000 Tonnen pro Jahr erfüllten sich jedoch nicht, während des Kriegs produzierten die Deutschen Fettsäure-Werke nur etwa 3000 Tonnen jährlich.[12]
Nachkriegszeit

Gegen Ende des Zweiten Weltkriegs und in den ersten Nachkriegsjahren verschlechterte sich die Versorgung der Bevölkerung mit Fett rapide. 1947 sank der jährliche Pro-Kopf-Verbrauch in Deutschland, der 1938 noch 25,6 Kilogramm betragen hatte, auf 5,7 Kilogramm ab.[23] Politiker und Industrielle versuchten, einen Fettimport anzukurbeln und die Freigabe für die Wiederaufnahme der Fischer-Tropsch-Synthese und das Imhausen-Prosch-Verfahren zu erwirken.[22] Die Produktionsstätten der Deutschen Fettsäure-Werke besaßen für die Herstellung eine betriebsbereite Kapazität von etwa 600 Tonnen synthetischer Butter pro Monat.[15] Dazu setzte die Verwendung synthetischer Fettsäuren für technische Zwecke die gleiche Menge nativer Fettsäuren für Ernährungszwecke frei. Pro Tonne Butter benötigte das Imhausen-Prosch-Verfahren sieben Tonnen Kohle; um dieselbe Menge Butter importieren zu können, war in der Nachkriegszeit etwa der Verkaufserlös der zehnfachen Menge Kohle am Weltmarkt notwendig.[22]
Zwar gelang es Arthur Imhausens Sohn, Karl-Heinz Imhausen, die Produktion von kohlestämmigen Seifen und Fetten in der Nachkriegszeit kurzzeitig wieder aufzunehmen, wobei das Werk Witten 1946 etwa 350 Tonnen synthetisches Fett pro Monat produzierte.[24] Das Interesse an der Paraffinoxidation und kohlestämmigen Fettsäuren endete aber in den 1950er Jahren mit dem Einsetzen des Wirtschaftswunders. Das plötzlich vorhandene Überangebot an Naturfetten auf dem Weltmarkt führte zu einem starken Preisverfall bei Fetten und Fettsäuren. Auf dem Tensidmarkt konkurrierten die kohlestämmigen Fettsäuren sowohl mit oleochemischen als auch petrochemischen Produkten wie den Ziegler-Alkoholen.[25] Während dadurch in der westlichen Welt das Verfahren nicht mehr konkurrenzfähig war und Fettsäuren kaum noch künstlich hergestellt wurden, wurden sie in der Sowjetunion und China weiterhin produziert. So wurden 1978 in der Sowjetunion und China über 500.000 Tonnen Fettsäuren durch Oxidation an Mangan-Katalysatoren aus Wachs und Kerosin gewonnen.[26]
Rohstoffe
Die in der Oxidation eingesetzten Paraffine besitzen eine Kohlenstoffkettenlänge von etwa 18 bis 30 Kohlenstoffatomen, entsprechend einem Schmelzbereich von 28 bis 66 °C und einem Siedebereich von 320 bis 460 °C.[27] Aus Paraffinen dieses Kettenlängenbereichs lassen sich die Zielprodukte, die Fettsäuren mit einer Kettenlänge von 10 bis 18 Kohlenstoffatomen, in größter Ausbeute gewinnen. Die Paraffine sollten zum Erlangen einer hohen Produktqualität möglichst geradkettig sein. Die aus verzweigten Fettsäuren hergestellte Seife hatte eine schlechte Waschwirkung und einen charakteristischen Geruch. Der Sauerstoff greift bei verzweigten Paraffinen außerdem bevorzugt am tertiären Wasserstoffatom an. Dabei entstehen zwar geradkettige Fettsäuren, jedoch mit einem überproportional großen Anteil an niedermolekularen Fettsäuren.[27]
Die Paraffine stammten hauptsächlich aus zwei Verfahren, der Hochdruckhydrierung von Braunkohle und deren Verschwelungsteeren sowie aus der Fischer-Tropsch-Synthese. Beim Tieftemperatur-Hochdruck-Hydrierverfahren entstanden geradkettige, so genannte TTH-Paraffine mit einem Verzweigungsgrad von etwa 10 bis 15 %.[27] Obwohl Braunkohle in großer Menge zur Verfügung stand, führten die Absatzprobleme der Nebenprodukte der Verschwelung nur zu einer relativ geringen Verfügbarkeit an geeigneten Paraffinen aus dieser Quelle.[27] Daneben eignen sich die bei der Harnstoff-Extraktiv-Kristallisation von Schmierölen anfallenden n-Alkane ebenfalls als Rohstoffe.[28]
Beim Niederdruckverfahren der Fischer-Tropsch-Synthese fiel Paraffingatsch mit der richtigen Kohlenstoffkettenlängenverteilung an. Besser geeignet war das Gatsch des Mitteldruckverfahrens, da es besonders geradkettig war, aber noch 15 bis 20 % kurzkettig verzweigte Paraffine aufwies. Diese eigneten sich aufgrund ihrer tertiären Wasserstoffatome, die einem oxidativen Angriff leichter zugänglich sind, besonders gut als Startmaterial für die Paraffinoxidation. Außerdem war die Ausbeute an Zielparaffinen im Mitteldruckverfahren etwa vier bis fünf Mal höher als beim Niederdruckverfahren.[27] Dafür war der Kettenlängenbereich zu höheren Kettenlängen verschoben, was eine destillative Vorbereitung des Gatschs erforderte.
Wegen ihrer inhibierenden Wirkung lag die Grenze für den Anteil an schwefelhaltigen Verbindungen und Phenolen bei 0,05 %. Eine Hydrierung des Rohstoffs entfernte störende Olefine und sauerstoffhaltige Verbindungen. Petroleum eignete sich wegen des Naphthengehalts, der zu zähflüssigen Fettsäuren minderer Waschqualität führte, kaum als Rohstoff für die Paraffinoxidation.[27] Langkettig verzweigte Paraffine, Olefine und Naphthene führten in der Oxidation außerdem zu höheren Anteilen an unerwünschten Hydroxycarbonsäuren.[14] Die Prüfung der Wachseignung geschah mittels einer Test-Oxidation.
Verfahren
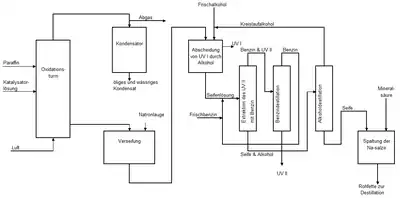
Das Verfahren bestand aus den drei Hauptschritten Oxidation, der Aufarbeitung des Oxidationsgemisches zu Rohfettsäuren und schließlich deren destillative Trennung in Fettsäurefraktionen.[29] Die chemische Industrie verarbeitete die Fettsäurefraktionen weiter zu Endprodukten wie Seifen, Waschmitteln, Weichmachern und synthetischem Fett.[29] Die Betreiber führten die Paraffinoxidation fast ausschließlich in einer Batch-Fahrweise durch, also diskontinuierlich.
Oxidation
Die Oxidation stellte eine wichtige Verfahrensstufe dar. Das Paraffin reagierte dazu im flüssigen Zustand bei möglichst niedrigen Temperaturen und in Gegenwart eines Katalysators etwa 15 bis 30 Stunden lang mit Luftsauerstoff, bis etwa 30 bis maximal 50 % des Paraffins in Fettsäure umgewandelt waren.[29] Dadurch minimierte sich die Bildung von unerwünschten Nebenprodukten, wie den in Petrolether unlöslichen Anteilen an Dicarbonsäuren, Hydroxycarbonsäuren und niedermolekularen Fettsäuren.[30]
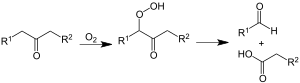
Nach einer kurzen, für autokatalytische Reaktionen typischen Latenzphase[31] sprang die Oxidation an, was an einer Wasserbildung sowie dem Anstieg der Säurezahl des Produkts festzustellen war. Enthielt das Rohmaterial Inhibitoren, mussten sie erst oxidiert werden, bevor die Reaktion ansprang. Die Anwesenheit von cyclischen und ungesättigten Kohlenwasserstoffen führte dagegen zur Bildung von Inhibitoren, die eine schon angesprungene Reaktion wieder unterdrückten oder zum Stillstand brachten.[30] Die Reaktion verlief schematisch nach der allgemeinen Gleichung:

Die entstandenen Fettsäuren unterlagen weiterhin der Nachoxidation, so dass gegenüber der statistisch zu erwartenden Verteilung eine übermäßige Menge an niedermolekularen Fettsäuren entstanden.[32] Als nicht-flüchtige Produkte entstanden Fettsäuren, Alkohole, Aldehyde, Ketone, Ester sowie Lactone, als flüchtige Produkte entstanden Kohlenstoffdioxid, Wasser, niedermolekulare Carbonsäuren und deren Ester, sowie Peroxide, Aldehyde und Alkohole.[30]
Druck und Temperatur
Ein hoher Reaktionsdruck erhöhte den Anteil des in Paraffin gelösten Sauerstoffs und damit die Reaktionsgeschwindigkeit. Eine Verdopplung der Drucks halbierte in etwa die Reaktionszeit.[33] In Magdeburg errichtete Hubbe & Fahrenholz während des Zweiten Weltkriegs eine Anlage, die bei einem Druck von 25 bar und ohne Katalysator arbeiten sollte.[14] Die Firma nahm die Anlage aber nicht mehr in Betrieb.[34] Alle anderen Anlagen arbeiteten unter Normaldruck. Der gelöste Sauerstoffanteil erhöhte sich über eine feinverteilte Einperlung der Luft, zum Beispiel über Filterkerzen oder Füllkörper.
Bei Temperaturen oberhalb von 170 bis 180 °C verlief die Oxidation recht schnell, aber es entstanden überoxidierte Produkte, die für eine Weiterverarbeitung zu Tensiden nicht geeignet waren.[35] Von Vorteil für die Gewinnung reiner Fettsäuren war eine relativ tiefe Oxidationstemperatur. Bei einer Umsatzbegrenzung auf etwa 30 % und bei Temperaturen von 105 bis 120 °C gewann die chemische Industrie Fettsäuren in hoher Selektivität und guter Qualität.[35] Die für industrielle Belange zu lange Reaktionszeit machte den Einsatz eines Katalysators notwendig.
Katalysator/Initiator

Die für Paraffinoxidation verwendeten Katalysatoren waren vielfältig. Oft handelte es sich um Oxide von Nebengruppenmetallen, etwa Cobaltsalze.[36] Als guter Initiator für die Paraffinoxidation erwies sich Kaliumpermanganat.[29][37] Alkalimetall-Salze agierten als Cokatalysator, die dem Verfahren über die Seifenanteile der aufgearbeiteten und rezyklierten Paraffine zugeführt wurden.
Eine Suspension des Kaliumpermanganats in den Paraffinen entstand durch Zugabe und schnelles Rühren einer konzentrierten wässrigen Permanganat-Lösung. Bei Temperaturen über 100 °C verdampfte das Wasser und es verblieb eine feinverteilte Suspension des Initiators. Die verwendete Menge betrug zirka 0,1 bis 0,3 % der eingesetzten Paraffinmenge und variierte mit der Qualität des Rohmaterials.[35] Durch die Zugabe des Katalysators erreichten die Betreiber bei relativ niedrigen Temperaturen von 110 bis 120 °C einen Zielumsatz von etwa 30 % nach 10 bis 15 Stunden Reaktionszeit.[38] Eine niedrige Reaktionstemperatur unterdrückte die Bildung unerwünschter Nebenprodukte wie Hydroxy- oder Dicarbonsäuren, die Produktfarbe verbesserte sich.
Verfahrensführung
Die Paraffinoxidation wurde in säurebeständigen Stahl- oder Aluminiumreaktoren durchgeführt. Diese besaßen einen Durchmesser von 1–3 Metern und eine Höhe von 8–12 Metern. Die Luft perlte über Filterplatten oder -kerzen ein, die Luft verteilte sich im Reaktor über Füllkörper. Die flüchtigen Säuren und Nebenprodukte lösten sich in mit Wasser befülltem Wäscher.[38] Die Oxidation einer Tonne Paraffin benötigte etwa 50 Kubikmeter Luft pro Stunde. Der Reaktionsstart erforderte anfangs eine Temperatur von etwa 150 °C. Nach dem Anspringen der Reaktion, nach etwa 20 bis 60 Minuten, wurde die Reaktionstemperatur auf unter 120 °C gesenkt.[38] Die überschüssige Luft transportierte niedermolekulare Anteile ab. Das Anspringen der Reaktion zeigte sich durch Wasseranfall in einem Kondensator, der auch die leichtflüchtigen Oxidationsprodukte auffing.[39]
Da die Oxidation exotherm war, musste der Reaktor gekühlt werden. Die freigesetzte Wärmemenge entsprach etwa 4,5 % der Verbrennungswärme des Paraffins, pro Tonne Oxidationsprodukt etwa 2100 Megajoule. War die Säurezahl von 70 erreicht, so bedeutete dies, dass der Zielumsatz erreicht war und die Oxidation abgebrochen werden konnte. Der Umsatz betrug dann etwa 30 %.[38] Die Umsatzbegrenzung reduzierte die Bildung von Fettsäurefolgeprodukten wie Hydroxycarbonsäuren und Dicarbonsäuren.[8]
Aufarbeitung der Rohfettsäuren
Die Rohfettsäuren enthielten eine Mischung von Carbonsäuren aller der im eingesetzten Paraffin enthaltenen Kettenlängen, nicht umgesetztes Paraffin sowie ein breites Spektrum von Oxidationsprodukten.[39] In der Aufarbeitung wurden die im Oxidationsprodukt enthaltenen Fettsäuren zuerst mit Wasser gewaschen, um die niedermolekularen sauren Bestandteile sowie den Katalysator abzutrennen.
Zur Abtrennung der Fettsäuren von den unverseifbaren Anteilen erfolgte zunächst eine Neutralisation des Rohoxidats mit Natronlauge unter Seifenbildung. Bei höherer Temperatur verseiften auch die entstandenen Ester. Die unverseifbaren Anteile wurden dann in einem Rührkessel mit 45-prozentigem Ethanol oder 20-prozentigem 2-Propanol gemischt. Die Seife löste sich in dem Alkohol, während die unverseifbaren Anteile sich als ölige Schicht auf dem Alkohol-Seifen-Gemisch absetzten.[39] Die ölige Schicht, die meist Paraffine enthielt und als Unverseifbares I (UV I) bezeichnet wurde, wurde in den Oxidationsprozess zurückgeführt. Der Anteil des UV I am Gesamtanteil unverseifbarer Komponenten betrug etwa 85 %.
Die restlichen unverseifbaren Anteile, als Unverseifbares II (UV II) bezeichnet, enthielt neben Resten von Paraffinen vor allem Oxidationsprodukte wie Alkohole, Ester und Lactone.[39] Durch Extraktion mit Benzin im Gegenstrom entfernten die Anlagenbetreiber diese Anteile. Getrennt wurden UV II und Benzin durch Destillation.[39] Nach der Trennung von Alkohol- und Seifenphase durch Destillation erfolgte im letzten Schritt die Rückgewinnung der Fettsäuren durch Ansäuern mit Mineralsäuren wie Schwefel- oder Salzsäure. Durch Waschen mit Wasser wurden Reste von kurzkettigen Carbonsäuren entfernt. Anschließend zerlegte eine Wasserdampfvakuumdestillation die Fettsäuren in einzelne Fraktionen.[29]
Produkte
Die Hauptlauffettsäuren mit einer Kohlenstoffkettenlänge von etwa 10 bis 20 Kohlenstoffatomen wurden mit Natronlauge zu Seifen neutralisiert. Im Gegensatz zu nativen Fettsäuren enthielten die synthetischen Fettsäuren Anteile an ungeraden Kohlenstoffketten.[8] Die Waschkraft der so erhaltenen Seifen unterschied sich nicht von den Seifen aus nativen Quellen. Die Anwesenheit von unverseifbaren Anteilen, die Gegenwart von Lactonen sowie die Anwesenheit von verzweigtkettigen Fettsäuren führte zum Teil zu Geruchsproblemen.[14] Eine Hydrierung reduzierte die Fettsäuren zu Fettalkoholen. Durch Sulfatierung mit Schwefeltrioxid entstanden aus den Fettalkoholen Fettalkoholsulfate.[8]
Durch Veresterung mit Glycerin stellte Imhausen als erster im großtechnischen Maßstab Fette und durch Emulgation später Butter aus den synthetischen Fettsäuren her.[8] Dafür verwendeten die Fettsäure-Werke nur natürliches Glycerin von ungenießbaren Fetten. Ein synthetisches Glycerin, das die IG Farben in Heydebreck herstellte, erwies sich als nicht rein genug für diesen Zweck.[14] Die Fettsäuren werden mit der stöchiometrischen Menge von Glycerin unter Verwendung von Zinkstaub verestert. Nach Beendigung der Reaktion löste eine Behandlung mit 20-prozentiger Schwefelsäure das Zink auf. Nach der Neutralisation mit Natronlauge wurden mit einem Gemisch aus Aktivkohle und Bleicherde Seifenspuren entfernt. Durch Vakuumdestillation erhielt Imhausen ein reines Fett, das zum Verzehr geeignet war.[14]
Die Vorlauffettsäuren wurden hauptsächlich zum Alkohol reduziert und nach Veresterung mit Phthalsäureanhydrid oder anderen Anhydriden zu Weichmachern verarbeitet. Die Nachlauffettsäuren mit höherer molarer Masse führten die Betreiber wieder in den Oxidationsprozess zurück.[8] Das Luftkühlerkondensat enthielt etwa 80 % Ameisensäure und 9 % Essigsäure und dienten zur Konservierung von Viehfutter. Die Lackindustrie verwendete den Rückstand der Fettsäuredestillation als Bindemittel für Pigmente.[40]
Mechanismus
Die ersten Ansatz zur Erklärung der Oxidation von paraffinischen Kohlenwasserstoffen lieferte die von Alexei Nikolajewitsch Bach und Carl Engler entwickelte Peroxidtheorie, die als Engler-Bach-Theorie bekannt ist.[41][42] Demnach bildet sich bei der Oxidation im ersten Schritt ein sekundäres Hydroperoxid. Diese Theorie, nach der sich dieses Hydroperoxid anschließend radikalisch zersetzt, bestätigten spätere Untersuchungen von Eric Rideal.[43]

Die Funktion des Metallkatalysators ist es, die Geschwindigkeit sowohl der Bildung als auch der Zersetzung dieses Hydroperoxid zu erhöhen. Dabei entsteht unter anderem ein Alkylradikal, das mit Sauerstoff zu einem Peroxoradikal reagiert. Dieses bildet durch Abstraktion eines Wasserstoffatoms aus einem weiteren Paraffinmolekül ein neues Alkylradikal sowie ein Hydroperoxid.[13]


Alkalisalze zersetzen ebenfalls die Peroxide und reagieren als Co-Katalysator. Die relative Geschwindigkeit des Sauerstoffangriffs auf primäre, sekundäre und tertiäre Wasserstoffatome beträgt etwa 1 : 3 : 33. Der Angriff an den hauptsächlich vorkommenden sekundären Wasserstoffatomen erfolgt rein statistisch und führt dazu, dass ein nahezu äquimolares Gemisch aller theoretisch möglichen Carbonsäuren gebildet wird.[30] Durch die Vielzahl der entstehenden Produkte gestaltete sich die genaue Aufklärung des Reaktionsmechanismus als schwierig. Die Reaktionsfolge der Oxidation und die Bildung der Haupt- und Nebenprodukte erklärt sich am besten nach dem von Wolfgang Langenbeck und Wilhelm Pritzkow entworfenen Schema:[44][45]
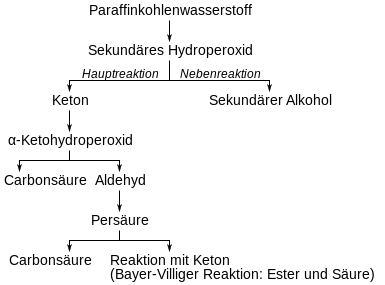
Als erste Stufe bildet sich ein Hydroperoxid, das als Hauptreaktion in Wasser und ein Keton zerfällt. Als Nebenreaktion entstehen sekundäre Alkohole gemäß folgendem Reaktionsschema:

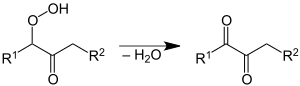
Das Keton unterliegt der Weiteroxidation. Bevorzugt wird dabei die Methylengruppe in α-Stellung zur Ketogruppe oxidiert. Das dabei entstehende α-Ketohydroperoxid zerfällt unter Umlagerung in einen Aldehyd und eine Carbonsäure.[46]
Pritzkow nutzte Cyclohexanon als Modellsubstanz und zeigte damit die Richtigkeit der Modellvorstellung.[47] Der Aldehyd geht über die Stufe der Peroxycarbonsäure und Weiterreaktion mit einem weiteren Aldehyd in die Carbonsäure über. Die Bildung der Carbonsäureester und weiterer Carbonsäuren erfolgt über die Baeyer-Villiger-Oxidation des Ketons mit Peroxycarbonsäure gemäß folgender Gleichung:[46]

Eine weitere Reaktionsmöglichkeit der α-Ketohydroperoxide ist die Bildung von Diketonen.
Diese reagieren mit Peroxycarbonsäuren unter Bildung von Säureanhydriden und Carbonsäuren.[46]

Bashkirov-Oxidation
Wird die Paraffinoxidation in Gegenwart von Borsäure durchgeführt, entstehen als Hauptprodukt sekundäre Fettalkohole. Dieses Verfahren wird nach seinem Entdecker Andrei Nikolajewitsch Bashkirov, der das Verfahren in den 1950er Jahren entwickelte, Bashkirov-Oxidation genannt.[48] Bei der Bashkirov-Oxidation kommt es nicht zu einem Abbau des ursprünglichen Paraffins, die entstehenden Alkohole entsprechen in ihrer Kettenlänge den als Rohmaterial eingesetzten Paraffinen.[49] Die Kettenlängen des Ursprungsparaffine sind im Vergleich zu den in der Paraffinoxidation eingesetzten dementsprechend kürzer.
Das Verfahren wird bei einer Temperatur von 150 bis 160 °C, einem Anteil von 0,1 % Kaliumpermanganat als Initiator und etwa 5 % Borsäure durchgeführt. Die Borsäure fängt die entstehenden Hydroperoxide ab und überführt sie in deren Ester, die unempfindlich gegen die weitere Oxidation sind. Durch anschließende Hydrolyse des Esters werden sekundäre Alkohole erhalten, die zu sekundären Alkylsulfaten oder mit Ethylenoxid zu nichtionischen Tensiden umgesetzt werden. Um Nebenreaktionen einzudämmen, wird der Umsatz auf etwa 20 % begrenzt, der Sauerstoffgehalt der Luft wird durch Verdünnung mit Stickstoff auf 3,5 % gesenkt.[50] Der Umsatz lässt sich bei diesem Verfahren infrarotanalytisch durch die Absorptionsintensität der Bor-Sauerstoff-Streckschwingung von Boratwachs überwachen.[51] Die Bashkirov-Oxidation findet in speziellen Verfahren Anwendung, etwa bei der Herstellung von Cyclododecanon. Dabei wird über die Oxidation von Cyclododecan mit Luftsauerstoff Cyclododecanol gewonnen, das zum Cyclododecanon dehydriert wird.[52]
Literatur
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956.
- Wilhelm Keim, Arno Behr, Günther Schmitt: Grundlagen der Industriellen Chemie. Otto Salle Verlag, 1985, ISBN 3-7935-5490-2.
- Klaus Weissermel, Hans-Jürgen Arpe: Industrielle Organische Chemie: Bedeutende Vor- und Zwischenprodukte. Wiley-VCH, 2007, ISBN 978-3-527-31540-6.
- Birgit Pelzer-Reith und Reinhold Reith: „Fett aus Kohle“? Die Speisefettsynthese in Deutschland 1933-1945. In: Technikgeschichte, Bd. 69 (2002), H. 3, S. 173–205.
Weblinks
- Welt im Film 121/1947 – 19. September 1947 (von 2:40 bis 4:20 min.), Fett aus Kohle, Wochenschau von 1947 über die Produktion von Butter und Seife bei den Deutschen Fettsäurewerken
Einzelnachweise
- Karl Freiherr von Reichenbach, Franz Wilhelm Schweigger-Seidel: Das Kreosot in chemischer, physischer und medicinischer Beziehung. 2., verm. Aufl., Leipzig, Barth 1835.
- P. Gotthard Hofstädter: Ueber künstliches und mineralisches Paraffin. In: Annalen der Chemie und Pharmacie. 91, 1854, S. 326–334, doi:10.1002/jlac.18540910307.
- C. H. Gill, Ed. Meusel: XLI. On paraffin and the products of its oxidation. In: Journal of the Chemical Society. 21, 1868, S. 466, doi:10.1039/JS8682100466.
- A. G. Pouchet: Einwirkung der Salpetersäure auf das Paraffin. In: Polytechnisches Journal. 214, 1874, S. 130–132.
- P. Bolley: Ueber einige neue Eigenschaften des Paraffins und über die Paraffinbäder. In: Polytechnisches Journal. 190, 1868, S. 121–124.
- C. Engler, J. Bock, Ber. dtsch. chem. Ges., 1879, 12, S. 2186.
- Patent US335962: Converting Petroleum and similar Hydrocarbons into Acids. Veröffentlicht am 9. Februar 1886, Erfinder: Eugen Schaal.
- Arthur Imhausen: Die Fettsäure-Synthese und ihre Bedeutung für die Sicherung der deutschen Fettversorgung. In: Kolloid-Zeitschrift. 103, 1943, S. 105–108, doi:10.1007/BF01502087.
- Adolf Grün: Die Oxydation von Paraffin. In: Berichte der deutschen chemischen Gesellschaft (A and B Series). 53, 1920, S. 987–996, doi:10.1002/cber.19200530618.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 491–493.
- Bernd Kaiser: Die Implikationen wirtschaftspolitischer Rahmenbedingungen für die Rohstoffbeschaffung internationaler Industrieunternehmen und sich hieraus ergebende Unternehmensstrategien am Beispiel der Henkel-Gruppe. Inaugural-Dissertation zur Erlangung des akademischen Grades eines Doktors der Wirtschafts- und Sozialwissenschaften (Dr. rer. pol.) der Friedrich-Alexander-Universität Erlangen-Nürnberg, 2009, S. 142–154. (PDF)
- Ralph Klein: Arthur Imhausen (1885–1951). In: Wolfgang Weber (Hrsg.): Ingenieure im Ruhrgebiet (= Rheinisch-Westfälische Wirtschaftsbiografien). Band 17. Aschendorfer Verlagsbuchhandlung GmbH, Münster 1999, ISBN 3-402-06753-6, S. 344–372.
- Karl T. Zilch: Synthetic fatty acids in Eastern Europe. In: Journal of the American Oil Chemists’ Society. 45, 1968, S. 11, doi:10.1007/BF02679036.
- Synthetic Fatty Acids and Detergents. In: B.I.O.S. Final Report No. 1722, Item No. 22. Archiviert vom am 2. April 2015; abgerufen am 14. März 2015.
- Birgit Pelzer-Reith, Reinhold Reith: "Fett aus Kohle"? Die Speisefettsynthese in Deutschland 1933–1945. In: Technikgeschichte, 69, 2002, S. 173–206.
- Michael Wildt: Der Traum vom Sattwerden. Hamburg 1986, ISBN 3-87975-379-2, S. 17.
- Bruno Fischer: Ruhrgebiet 1933–1945. Der historische Reiseführer. Ch. Links Verlag, 2009, ISBN 978-3-86153-552-2, S. 86.
- Berend Strahlmann: Imhausen, Arthur. In: Neue Deutsche Biographie (NDB). Band 10, Duncker & Humblot, Berlin 1974, ISBN 3-428-00191-5, S. 145 f. (Digitalisat).
- Richard Breitman: Der Architekt der „Endlösung“. Heinrich Himmler und die Vernichtung der europäischen Juden. Paderborn 1996, S. 52–53.
- H. Kraut: The Physiological Value of Synthetic Fats. In: British Journal of Nutrition. 3, 1949, S. 355, doi:10.1079/BJN19490049.
- Hans Heinrich Meyer-Döring: Ist die Verwendung synthetischer Fettsäuren für Speisezwecke ungefährlich? In: Klinische Wochenschrift. 27, 1949, S. 113, doi:10.1007/BF01471275.
- Zwanzig Minuten Kohlenklau, dafür doppelte Fettration. In: Der Spiegel. Nr. 49, 1947, S. 6 (online).
- W. Heimann: Fette und Lipoide, Springer Verlag, 1969, ISBN 978-3-642-46190-3, S. 7.
- J. W. Vincent: Aspects of the Synthetic Fatty Acid and Synthetic Fat Industries in Germany. In: B.I.O.S. Final Rept. 805, 1946. 1946, archiviert vom am 2. April 2015; abgerufen am 18. März 2015.
- Siegeszug der Erdölmänner. In: Der Spiegel. Nr. 31, 1952, S. 10–11 (online).
- H. Fineberg: Synthetic fatty acids. In: Journal of the American Oil Chemistry Society. 56, 1979, S. 805A, doi:10.1007/BF02667451.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 493–497.
- Wilhelm Keim, Arno Behr und Günter Schmitt: Grundlagen der Industriellen Chemie. Technische Produkte und Prozesse, Otto Salle Verlag, 1985, ISBN 3-7935-5490-2, S. 250.
- G. Wietzel: Herstellung synthetischer Fettsäuren durch Oxydation von paraffinischen Kohlenwasserstoffen mit molekularem Sauerstoff. In: Chemical Engineering Science. 3, 1954, S. 17–IN4, doi:10.1016/S0009-2509(54)80003-0.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 498–499.
- Francis Francis, Walter Frederick Millard u. a.: XLVI.- The velocity of oxidation of paraffin wax. Parts I-IV. In: Journal of the Chemical Society, Transactions. 125, 1924, S. 381, doi:10.1039/CT9242500381.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 478–490.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 502–503.
- Patent DE739570: Verfahren zur Herstellung von Oxydationserzeugnissen, wie Fettsäuren oder Alkoholen, durch Oxydation von Kohlenwasserstoffen. Veröffentlicht am 12. August 1943, Erfinder: Karl Blass.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 500–502.
- Patent US2249708: Method of oxidation of paraffin. Veröffentlicht am 15. Juli 1941, Erfinder: Mildred M. Hicks-Bruun.
- N. M. Emanuel': The Oxidation of Hydrocarbons in the Liquid Phase. Elsevier, 2013, ISBN 978-1-4831-4925-7, S. 324 (eingeschränkte Vorschau in der Google-Buchsuche).
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 504–508.
- Friedrich Asinger: Chemie und Technologie der Paraffinkohlenwasserstoffe. Akademie Verlag, 1956, S. 508–513.
- Ludwig Mannes: Über die Nebenprodukte der Paraffin-Oxydation. In: Die Chemie. 57, 1944, S. 6, doi:10.1002/ange.19440570102.
- C. Engler, W. Wild: Ueber die sogenannte, Activirung des Sauerstoffs und über Superoxydbildung. In: Berichte der deutschen chemischen Gesellschaft. 30, 1897, S. 1669, doi:10.1002/cber.18970300295.
- A. N. Bach: On the role of peroxides in the processes of slow oxidation. In: Zh. Russ. Phys-Chem. Soc., 29, 1897, S. 373–395.
- P. George, E. K. Rideal, A. Robertson: The Oxidation of Liquid Hydrocarbons. I. The Chain Formation of Hydroperoxides and Their Decomposition. In: Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 185, 1946, S. 288, doi:10.1098/rspa.1946.0019.
- W. Langenbeck, W. Pritzkow: Untersuchungen über den Mechanismus der Paraffinoxydation, 1. Teil. In: Fette, Seifen, Anstrichmittel. 55, 1953, S. 435, doi:10.1002/lipi.19530550704.
- W. Langenbeck, W. Pritzkow: Untersuchungen über den Mechanismus der Paraffinoxydation, 2. Teil. In: Fette, Seifen, Anstrichmittel. 55, 1953, S. 506, doi:10.1002/lipi.19530550805.
- Mihaly Freund, Gyula Mozes: Paraffin Products: Properties, Technologies, Applications. Elsevier Science Ltd., 1983, ISBN 978-0-444-99712-8, S. 64.
- Wilhelm Pritzkow: Über die Autoxydation von Ketonen, I. Mitteil.: Die Oxydation von Cyclohexanon. In: Chemische Berichte. 87, 1954, S. 1668, doi:10.1002/cber.19540871110.
- A. N. Bashkirov, V. Kamzolkin: Synthesis of Higher Aliphatic Alcohols by Direct Oxidation of Paraffinic Hydrocarbons. In: Proc. World Pet. Cong., 4, 1959, S. 175–183 (Abstract).
- F. Novak, V. Kamzolkin, Y. Talyzenkov, A. Bashkirov: Mechanism of the effect of boric acid on liquid-phase oxidation of paraffin hydrocarbons. In: Petroleum Chemistry U.S.S.R. 7, 1967, S. 59–64, doi:10.1016/0031-6458(67)90010-X.
- Wilhelm Keim, Arno Behr und Günter Schmitt: Grundlagen der Industriellen Chemie. Technische Produkte und Prozesse, Otto Salle Verlag, 1985, ISBN 3-7935-5490-2, S. 124–125.
- W. Zenker: Infrared Method for the Direct Control of Paraffin-Wax Oxidation in the Presence of Boric Acid. In: Fette, Seifen, Anstrichmittel. 77, 1975, S. 221–224, doi:10.1002/lipi.19750770605.
- K. Weissermel, H.-J. Arpe: Industrielle Organische Chemie: Bedeutende Vor- und Zwischenprodukte. Wiley-VCH, 2007, ISBN 978-3-527-31540-6, S. 243–244.