Pharmazeutische Technologie
Die pharmazeutische Technologie ist ein wissenschaftlich-technischer Fachbereich, der sich mit der Herstellung von Arzneimitteln befasst. Ein wesentlicher Teilbereich der pharmazeutischen Technologie ist die Arzneiformenlehre – Arzneiformen sind Zubereitungen der Arzneistoffe in einer bestimmten Form, zum Beispiel als Tabletten.

Eine ältere, bis in die 1960er Jahre übliche Bezeichnung für die Zubereitung und Herstellung von Arzneimitteln ist Galenik (benannt nach dem griechischen Arzt Galenos). Die Bezeichnung Galenik wird bei der traditionellen, handwerklichen Herstellung von Arzneimitteln ebenso verwendet wie in modernen Pharmaunternehmen. Der Begriff pharmazeutische Technologie bezieht sich in der Regel auf die moderne Arzneimittelproduktion.
Die Approbationsordnung für Apotheker vom 23. August 1971, welche für das Pharmaziestudium eine um die industriell-maschinelle Komponente der galenischen Technik erweiterte Ausbildung vorsah, hatte die Begrifflichkeiten Arzneiformenlehre bzw. Pharmazeutische Technologie eingeführt.[1]
Arzneiform
Als Arzneiform oder Darreichungsform bezeichnet man die Zubereitung, in der ein Wirkstoff appliziert wird. Im einfachsten Fall des einzeln abgeteilten Pulvers ohne Hilfsstoffe, stellt der Wirkstoff selbst schon die vollständige Arzneiform dar. Allerdings haben einzeln dosierte Pulver, sei es als reiner Wirkstoff oder als Gemisch aus Wirk- und Hilfsstoffen, als eigenständige Arzneiform aufgrund der vielen Nachteile heute kaum noch Bedeutung. Eine Arzneiform besteht demnach aus Wirkstoffen und Hilfsstoffen, die in einer besonderen Art verarbeitet sind.
Der Arzneiform kommt – neben dem eigentlichen Wirkstoff oder Wirkstoffgemisch – eine entscheidende Bedeutung für die Wirksamkeit des Arzneimittels zu. Sie bestimmt die wesentlichen Eigenschaften der fertigen pharmazeutischen Zubereitung (Herstellung, Lagerung, Haltbarkeit, Pharmakokinetik, mikrobielle Reinheit, Verpackung usw.) mit. Um die Wirkung eines Arzneimittels richtig zu beurteilen, muss die Arzneiform neben dem reinen Wirkstoff stets berücksichtigt werden.
Forderungen an eine Arzneiform
An eine Arzneiform werden viele verschiedene Anforderungen gestellt:
- Dosierungsgenauigkeit
- chemische Stabilität
- biologische Stabilität
- physikalische Stabilität
- Gleichförmigkeit
- physiologische Verträglichkeit
- Wirkstoffgehalt und Wirkstoffverteilung
- Äußere Form
- Aussehen
- Kaschieren eines schlechten Geruches, Geschmacks
Entwicklung einer Arzneiform
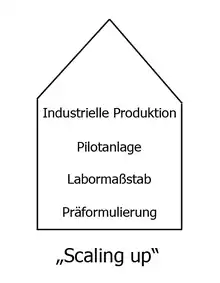
Anfänglich entwickelten sich Arzneiformen empirisch, das heißt durch einfaches Ausprobieren. Was nützlich war, wurde beibehalten. So kam es zu den „historischen“ Arzneiformen wie der Pille, die es heute – außer dem Namen – nicht mehr gibt. (Nebenbei: Was heute landläufig als „Pille“ bezeichnet wird – also die oralen Kontrazeptiva – sind in den meisten Fällen Dragees bzw. Filmtabletten.)
Heute werden Arzneiformen aufgrund wissenschaftlicher Erkenntnisse gezielt entwickelt. Am Anfang steht die „Präformulierung“ vergleichbar einem Prototyp. Die nächste Stufe besteht darin, die Arzneiform in kleinem Maßstab zu reproduzieren: „Labormaßstab“. Danach steht die erste Versuchsanlage und am Ende des Prozesses die großtechnische Herstellung. Dieser Vorgang wird als „Scaling-up“ bezeichnet. Auf jeder „Vergrößerungsstufe“ treten spezifische Probleme auf: Eine Tablette im Labor herzustellen oder in großer Menge auf Rundläuferpressen ist eben ein Unterschied.
Präformulierungsuntersuchungen
Von großer Bedeutung ist die Löslichkeit des Wirkstoffes in wässrigem Milieu. Bei einer Löslichkeit von weniger als 0,5 % kann die Resorption sehr schlecht sein, jedoch ist manchmal auch eine sehr hohe Löslichkeit schlecht. Durch Übergang des Wirkmoleküls vom Magen-Darm-Trakt ändert sich der pH-Wert erheblich. Durch Bestimmung des pKa einer Substanz lässt sich die Löslichkeit des Stoffes im Wasser berechnen. Wirkstoffe können in unterschiedlichen Zustandsformen auftreten (Polymorphie), die sich durch unterschiedliche Löslichkeit und Stabilität unterscheiden.
Beeinflussung der Löslichkeit
Sind aufgrund von pH-Abhängigkeiten, Polymorphieuntersuchungen gute Wirkstoffe vermutlich schlecht resorbierbar, so kann der Galeniker physikalisch-chemische und chemische Maßnahmen zur Verbesserung der Löslichkeit eines Wirkstoffes anwenden. Bei den physikalisch-chemischen Verfahren packt man den Wirkstoff häufig in eine hydratfreie Umgebung (zum Beispiel in eine Polyethylenglykol-Matrix). Bei Auflösung der festen Lösung, der Matrix, wird der Wirkstoff dispergiert und kann dann solvatisiert werden. Eine weitere Methode ist die Erwärmung des Lösungsmittels, damit sich mehr Stoff lösen lässt.
Durch Einführung von Substituenten, die durch enzymatische Einwirkung abgespalten werden können, werden Wirkstoffe chemisch verändert. Dies dient zur Verbesserung der Löslichkeitseigenschaften, zur verbesserten Überwindung der Blut-Hirn-Schranke, zur Verlängerung der Wirkung einer Substanz, zur Verringerung des schlechten Geschmacks.
Der Wirkstoff mit Substituent wird als Pro-Drug bezeichnet. Ein Beispiel für ein Pro-Drug ist das Phenacetin, das durch Acetat-Abspaltung in Paracetamol überführt wird. Zur Verbesserung der Wasserlöslichkeit wurde bei Methylprednisolon zum Beispiel Bernsteinsäure eingeführt.
Bei Vorliegen von Säuren und Basen als Wirkstoffe können mitunter auch Salze eingesetzt werden.
Hilfsstoffe
Die verwendeten pharmazeutischen Hilfsstoffe dürfen nicht mit dem Wirkstoff reagieren. Toxikologisch darf von den Hilfsstoffen keine Gefahr ausgehen. Zur Bestimmung der Stabilität eines Wirkstoffes zusammen mit Hilfsstoffen wird das Factorial Design genutzt. Hierbei untersucht man einen Wirkstoff mit verschiedenen Hilfsstoffen in Abhängigkeit von der Zeit. Die Wirkstoffe werden längere Zeit bei verschiedenen Temperaturen und Feuchtigkeitsgraden gelagert. Die Konzentrationsabnahme des Wirkstoffes gibt Hinweise zur Stabilität des Wirkstoffes in einem Hilfsstoff.
Good Manufacturing Practice (GMP)

Die wörtliche Übersetzung von Good Manufacturing Practice ist „Gute Herstellungspraxis“ (auch ‚Gute Manieren beim Produzieren‘), von den produzierenden Firmen und den Betroffenen wird dies meist mit „Give more paper“ (dt. ‚Große Mengen Papier‘) wiedergegeben.
Anlass zur Einführung einer allgemeinen Richtlinie zur Herstellungspraxis war der Wunsch, allgemein anerkannte Methoden ordentlicher pharmazeutischer Fertigung international festzulegen, um Unbedenklichkeit und Sicherheit von Arzneimitteln, also die pharmazeutische Qualität, über die gesamte Laufzeit eines Arzneimittels zu gewährleisten.
Die Laufzeit beginnt mit der Gewinnung und den ersten Verarbeitungsschritten der Ausgangsstoffe (Wirk- und Hilfsstoffe) und endet mit dem Ablauf des Verfallsdatums auf dem Fertigarzneimittel. Im Allgemeinen umfasst diese Zeitspanne etwa 3–5 Jahre und hängt von der Art der benutzten Stoffe, der Arzneiform und der Dauer der Zwischenlagerung sowie der Laufzeit des fertigen Arzneimittels ab.
1986 führte die WHO eine zunächst unverbindliche Richtlinie ein, die 1992 neu gefasst und modernisiert wurde. In der 1992er-Fassung fand besonders die „In-Prozess-Kontrolle“, also das genaue Einhalten und Überprüfen der einzelnen Herstellungsschritte, starke Berücksichtigung. Grund dafür ist die Einsicht, dass sich Qualität nicht durch nachträgliches Prüfen und Kontrollieren erreichen lässt, sondern nur durch ordentliches Herstellen und die Qualitätskontrolle – im Gegensatz zu früher – die produzierte Güte nur noch abschließend feststellt.
1970 wurde die Pharmaceutical Inspection Convention (PIC) („Übereinkunft zur Prüfung von Arzneimitteln“), eine Richtlinie zum ordnungsgemäßen Prüfen von Arzneimitteln, verabschiedet.
Seither hat auch die EU eine eigene Richtlinie (EG-GMP) verabschiedet, die in etwa den Vorschriften der PIC entspricht. Sie besteht aus einer verbindlichen Richtlinie und einem empfohlenen Teil (Leitfaden).
Erwähnenswert sind noch die SOP (Standard Operating Procedures), in denen die einzelnen Herstellungsschritte genau dokumentiert sind. Sie garantieren eine durchgängige Transparenz des gesamten Produktionsprozesses.
In anderen Bereichen der Pharmazie werden diese Regelwerke durch entsprechende Richtlinien ergänzt:
- GLP – Good Laboratory Practice
- GCP – Good Clinical Practice
- GAMP – Good Automated Manufacturing Practice
Die GMP und verwandte Richtlinien decken unter anderen diese Themenkreise ab:
- Sorgfaltspflichten
- Ausbildung des Personals
- Räumlichkeiten
- Trennung von Herstellung, Verpackung und Lagerung
- Prüfung
- Kennzeichnung
- Hygiene, besonders mikrobielle Kontamination
- Qualität der Materialien
- Regeln zur Selbstinspektion und zum Audit (Fremdkontrolle)
- Prozessinterne Kontrolle
- Validierung
- Qualitätskontrolle
Literatur
- Alfred Fahr: Pharmazeutische Technologie. Für Studium und Beruf. Deutscher Apotheker Verlag, Stuttgart 2015, 12. neu bearbeitete Auflage, ISBN 978-3-7692-6194-3. Ursprünglich entwickelt von Rudolf Voigt.
- David Schoebel: Multikriterielle Gestaltung von pharmazeutischen Wirkstoffanlagen. Gabler, Betriebswirtschaftlicher Verlag (2008), ISBN 3834912166
Einzelnachweise
- Rudolf Schmitz: Der Arzneimittelbegriff der Renaissance. In: Rudolf Schmitz, Gundolf Keil: Humanismus und Medizin. Acta humaniora, Weinheim 1984 (= Deutsche Forschungsgemeinschaft: Mitteilungen der Kommission für Humanismusforschung. Band 11), ISBN 3-527-17011-1, S. 1–21, hier: S. 18.