Milzbrandtoxin
Milzbrandtoxin (auch: Anthrax-Toxin) ist ein Proteingemisch, das vom Milzbrand-Erreger, dem Bakterium Bacillus anthracis produziert wird und das verantwortlich für die Gefährlichkeit einer Milzbrandinfektion ist. Es wird vom Bakterium während der Infektion ausgeschieden, dringt in die Zellen des Wirts ein und verursacht ihre Zerstörung.
Die Untereinheiten des Milzbrandtoxins heißen Protektives Antigen (PA), Letalfaktor (LF) und Ödemfaktor (EF). Die sich aus dem Milzbrandtoxin bildenden Proteinkomplexe gehören zur großen Gruppe der Bacillus- und Clostridium-Exotoxine, die aus zwei sich funktionell ergänzenden Untereinheiten aufgebaut sind (AB-Toxine): PA+LF ist das Letaltoxin und PA+EF das Ödemtoxin.[1][2]
Für die Strukturbiologie ist Milzbrandtoxin ein besonders zugängliches Modell, um die Mechanismen beim Transmembrantransport großer Proteine zu untersuchen. Obwohl die Einzelheiten des Vergiftungsprozesses durch Milzbrandtoxin ausgiebig untersucht wurden, sind aufgrund der Komplexität des Vergiftungsgeschehens noch nicht alle Fragen beantwortet. Trotzdem stehen neben Antibiotika mehrere Impfungen und hochwirksame Antikörper für die Behandlung zur Verfügung.[3]
Geschichte
Milzbrand war die erste Krankheit, bei der als Ursache die Infektion mit Mikroorganismen zweifelsfrei nachgewiesen werden konnte. Robert Koch gelang es nicht nur, im Jahr 1876 den Krankheitserreger (Bacillus anthracis) im Labor zu vermehren, er wies außerdem dessen Sporenbildung nach und konnte gesunde Tiere damit krank machen. Dieser erste Nachweis eines Übertragungswegs leitete das so genannte goldene Zeitalter der Mikrobiologie ein.[4] Nur wenige Jahre später entwickelte Louis Pasteur einen Impfstoff für Schafe, nachdem er zuvor mit Geflügelcholera-Bakterien (Pasteurella multocida) das Prinzip der Abschwächung der Virulenz (Attenuierung) durch Kultur bei erhöhter Temperatur demonstriert hatte. Die historischen Feldversuche fanden 1881 in Pouilly-le-Fort statt.[5][6]
Die Menge und Qualität der Veröffentlichungen über Milzbrandtoxin in den Jahren 1953 bis 1968 ist bemerkenswert, im Gegensatz zum fast völligen Fehlen derselben zwischen 1969 und den frühen 1980er Jahren. Im Jahr 1954 wurde erstmals gezeigt, dass das Blutserum infizierter und spät mit Antibiotika behandelter Meerschweinchen Toxine enthält. In den Jahren bis 1962 entwickelten britische und amerikanische Forscher die Methoden zur Isolierung und Reinigung der Untereinheiten LF, EF und PA. Als klar wurde, dass das Ödemtoxin (s. u.) nicht tödlich ist, konzentrierte man sich ausschließlich auf die Untersuchung des Letaltoxins. Die Entdeckung im Jahr 1962, dass ein spezieller Laborratten-Stamm, die so genannte Fischer-Ratte, hochsensibel auf das Toxin reagiert, wird auch heute noch zur Messung der Reinheit einer Toxinpräparation benutzt.[7][8][9]
Vergiftungsprozess

Sind die Lebensbedingungen für B. anthracis günstig, bilden sich aus den Endosporen wieder Bakterien; dies geschieht beispielsweise auf der Haut, in der Lunge oder im Darm von Wirbeltieren, wobei die Proteine LF, PA und EF in großen Mengen produziert und sezerniert werden. Außerhalb des Bakteriums kürzt eine Protease das PA zu PA-63 und dieses bildet an der Wirtszelle eine Präpore, die maximal vier Moleküle LF und/oder EF binden kann. Nach der Einschleusung des Toxins in die Wirtszelle ist es Bestandteil eines Endosoms, das innerhalb von Minuten zum Lysosom reift, dadurch werden die Proteine im Innern einer starken Ansäuerung ausgesetzt. Dies führt bei der Präpore zur Bildung einer „Klammer“, die aus dem Ring ein Porenbildendes Toxin macht, die die Membran durchdringt und worüber die gebundenen LF- und EF-Einheiten ins Zytosol gelangen.[10][11]
Sind LF und EF frei im Zytosol, können ihre disruptiven Funktionen nicht gestoppt werden. Aufgrund ihrer Enzymeigenschaft werden sie dabei auch nicht verbraucht. LF ist eine Protease, die insbesondere in den MAPKK-Signalweg eingreift, während EF als Adenylylcyclase ein Übermaß an cAMP produziert und die Überexpression der Milzbrandtoxin-Rezeptoren veranlasst. Die Eskalation durch große Toxinmengen führt zum Zelltod.
Da alle Zellen sensibel gegenüber Milzbrandtoxin sind und das Absterben der Zellen des angeborenen Immunsystems die Infektion begünstigt, werden als Teil der Immunantwort immer größere Mengen Interleukine ausgeschüttet. Der dadurch resultierende Schock mit Atmungs- und Herzversagen ist letztlich die Todesursache für den Wirt. Wird das Toxin direkt appliziert, verläuft das Krankheitsbild anders als bei einer Infektion. Hier ist kein systemischer Schock, sondern die Zerstörung von Blutgefäßen die letztliche Todesursache. Außerdem haben einzelne Wirbeltierarten (Nagetiere, Frösche) eine besondere Sensibilität auf das Toxin und können innerhalb weniger Stunden daran sterben, während eine letale Infektion Tage beanspruchen kann.[1][7]
Die Untereinheiten PA, LF, EF
Die Pathogenität des Milzbrandbakteriums ist hauptsächlich mit der Anwesenheit der beiden Plasmide pXO1 und pXO2 verknüpft, die 110 und 60 Megadalton schwer sind. Während pXO2 unter anderem für die Polyglutamathülle des Bakteriums codiert, enthält pXO1 den genetischen Code für die Toxin-Untereinheiten als Teil einer so genannten Pathogenitätsinsel, die sich zwischen den Positionen 117177 und 162013 (gesamt im Plasmid 181654 Basenpaare) erstreckt und acht identifizierte Gene enthält. Drei davon, die Gene pagA, lef und cya codieren für die Proteine PA, LF und EF. Sobald ein Schwellenwert an Carbonsäuren oder Kohlenstoffdioxid überschritten wird, reagiert der Transkriptionsfaktor atxA (Teil eines noch unbekannten Zweikomponentensystems) und startet die Expression der Pathogenitätsfaktoren Toxin plus Polyglutamat. Die Untereinheiten PA, LF und EF werden im Verhältnis 20:5:1 ausgeschüttet.[7][12][13]
Protektives Antigen (PA)
| Protektives Antigen (PA-63) | ||
|---|---|---|
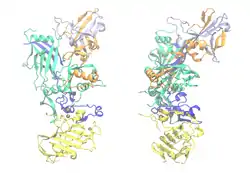
| ||
| Bändermodell von PA-83, die einzelnen Domänen gefärbt, Calcium als rote Kugeln: Gelb. PA-20, wird abgeschnitten. Blau. Calcium- und LF/EF-bindend. Grün. Membrandomäne. Orange. Oligomerisierungs-Schnittstelle. Pink. Rezeptor-bindend (nach PDB 1ACC) | ||
| Masse/Länge Primärstruktur | 568 aa | |
| Sekundär- bis Quartärstruktur | Homooctamer | |
| Kofaktor | Ca++ | |
| Präkursor | prepro-PA (764 aa) PA-83 (735 aa) | |
| Bezeichner | ||
| Gen-Name(n) | pagA | |
| Externe IDs | ||
| Transporter-Klassifikation | ||
| TCDB | 1.C.42 | |
| Bezeichnung | BAPA-Familie | |
| Vorkommen | ||
| Homologie-Familie | Hovergen | |


Das protektive Antigen (PA) ist ein Protein, das in einer Länge von 735 Aminosäuren von B. anthracis ausgeschieden wird. In der verkürzten Form (PA-63; 568 Aminosäuren) ist es Bestandteil des Milzbrandtoxins und lagert sich mit sieben weiteren Einheiten PA-63 zur achtteiligen Präpore zusammen. Diese Präpore bindet eine oder mehrere der LF/EF-Untereinheiten, veranlasst durch Bindung an einen Wirtsrezeptor die Einschleusung in eine Wirtszelle mittels Endozytose, und wandelt sich schließlich innerhalb des Endosoms zur ausgewachsenen Pore, welche die Ausschleusung der LF/EF-Untereinheiten aus dem Endosom ins Zellinnere (Zytosol) bewirkt. Diese Transportfunktion von PA-63 ist essenziell für die Pathogenität des Bakteriums.[14]
Die Transportgleichung der Pore lautet:
- LF oder EF (Endosom-Innenraum)
LF oder EF (Zytosol)

Expression, Modifikation, Oligomerisierung
Das auf dem Plasmid pXO1 positionierte pagA-Gen (2294 Basenpaare) codiert für ein 764 Aminosäuren langes Vorläuferprotein, das eine 29 Aminosäuren lange Signalsequenz am Translationsanfang enthält. Das Sec-Transportsystem in der Zellmembran des Bakteriums erkennt diese Signalsequenz und veranlasst daraufhin die Sekretion des noch ungefalteten Proteins, wobei die Signalsequenz abgetrennt wird. Das extrazelluläre Chaperon prsA hilft dem nunmehr 735 Aminosäure langen PA-83 bei der Faltung in die endgültige Form.
Darüber, wo genau im Wirt das PA-83 zum PA-63 gekürzt wird, gibt es widersprüchliche Ergebnisse. In jedem Fall spaltet eine Protease weitere 167 Aminosäuren von PA-83 ab. Erst diese Kürzung legt die Proteindomänen frei, die es den Faktoren LF und EF erlauben, an PA-63 zu binden. Und erst jetzt ist PA-63 in der Lage, sich mit mehreren gleichen Einheiten zu einer ringförmigen Präpore zusammenzulagern. Obwohl noch unklar ist, ob die bevorzugte Form von PA-63 das Heptamer oder Octamer ist, scheint das Octamer bei den Bedingungen, die bei einer Infektion herrschen, stabiler als das Heptamer zu sein. Gleichwohl können beide Formen die Endozytose an Wirtszellen-Rezeptoren veranlassen.[14]
Bindung an Wirtsrezeptoren
Von drei Rezeptorproteinen, die alle auf Wirbeltierzellen vorkommen, ist nachgewiesen, dass sie dem Milzbrandtoxin die Einschleusung in die Zelle erlauben. Es handelt sich um den Milzbrandtoxinrezeptor 1 (ATR, auch: TEM8), der normalerweise Typ-1-Kollagene bindet; den Milzbrandtoxinrezeptor 2 (ATR2, auch: CMG-2), der Laminin und Typ-4-Kollagene als natürliche Liganden hat; und heterodimeres Integrin-β1. Alle diese Rezeptoren enthalten eine so genannte Integrin-I- oder Von-Willebrand-A-Domäne, deren Aminosäuren ein zweiwertiges Metall-Ion (Ca++, Mn++, Mg++) auf der Rezeptoroberfläche komplexieren, und so eine Metallionen-abhängige Klebestelle bilden (MIDAS, von engl. metal-ion dependent adhesion site). Spätestens hier, an den Rezeptor gebunden, zerschneidet eine Protease das PA-83 und oligomerisiert das resultierende PA-63 zur sieben- oder achtteiligen Präpore, was zu einer Anhäufung von ebenso vielen Rezeptoren in der Zellmembran führt. An die Präpore wiederum sind bis zu vier LF- oder EF-Einheiten gebunden. Die Endozytose des Gesamtkomplexes, der nun bis zu 25 Untereinheiten enthält, erfolgt über cholesterinreiche Flecken der Membran, die Lipid Rafts, und wird durch Clathrin unterstützt. Aus den kristallographischen Daten ergeben sich die Abmessungen der Präpore: Durchmesser 160 Å, Höhe 85 Å und ein Innendurchmesser von etwa 35 Å.[10][14][15]
Konfigurationsänderung zur Pore

Aufgrund der generellen Schwierigkeit, Membrantransportproteine röntgenkristallografisch zu untersuchen, ist kaum etwas über die Struktur von Poren und ihren Bildungs- und Transportmechanismus bekannt. In dieser Hinsicht ist PA-63 eines der bestuntersuchten Transportproteine für den Transport großer Proteine. Zunächst wiesen Blaustein und Kollegen nach, dass PA-63 in künstlichen Membranen unter sauren Bedingungen einen Kanal bildet, der für Ionen durchlässig war. Spätere Untersuchungen mit verschieden großen Ionen ergaben für den Innendurchmesser einen Wert von 12 Å, ganz ähnlich dem anderer A/B-Toxine wie Botulinus-, Diphtherie- oder Tetanustoxin. Sobald die Pore einen der Faktoren LF oder EF transportierte, war sie für keine Ionen mehr durchlässig.[13]
Im Jahr 2004 gelang es Tam Luong Nguyen, durch Vergleich mit dem α-Hämolysin von Staphylococcus aureus, einen plausiblen Mechanismus für die Bildung der PA-63-Pore zu postulieren und ein räumliches Modell der Pore anzufertigen, das gut mit allen bis dahin vorhandenen Ergebnissen übereinstimmte. Essenziell ist dabei eine Proteindomäne, deren Rückgrat ein Mäandermotiv bildet, das im sauren Milieu wie eine Klammer nach außen klappt und ein beta-Faltblatt bildet. Sieben oder acht dieser Faltblätter formieren sich zur Röhre (beta-Fass), das genau die geforderten Eigenschaften besitzt. In einer wegweisenden Studie konnten Hiroo Katayama und Mitarbeiter im Jahr 2008 zeigen, dass das Chaperon GroEL geeignet ist, die PA-63-Pore so zu stabilisieren, dass elektronenmikroskopische Messungen durchgeführt werden können. Diese bestätigten im Nachhinein die Vermutungen des Nguyen-Modells.[3][16][17]
Translokation von LF und EF
Auch der Mechanismus des Transports der Virulenzfaktoren aus dem Endosom ist noch nicht vollständig geklärt. Große Fortschritte ergaben sich durch den Einsatz von elektrophysiologischen Messsystemen, die von mehreren Forscherteams ab 2004 entwickelt wurden. Diese bestehen aus zwei mit Kaliumchloridlösung gefüllten Kammern, die über eine ebene Phospholipid-Doppelmembran verbunden sind. Elektrische Spannungen und pH-Gradienten sind leicht einstellbar und messbar. Damit kann die Transportfähigkeit von gezielt an einzelnen Aminosäuren veränderten PA-63-Poren bzw. Faktoren unter realistischen Bedingungen gemessen und so festgestellt werden, welche Aminosäuren überhaupt an dem Prozess teilnehmen. Anhand solcher Versuche konnten ab dem Jahr 1994 verschiedene Forschergruppen belegen, dass LF mit dem N-Terminus zuerst durch die Pore transportiert wird; dass das elektrische Potenzial in Transportrichtung positiv ist; dass, wenn LF sich in der Pore befindet, kein Durchkommen für weitere Teilchen möglich ist; dass eine bestimmte Aminosäure, Phenylalanin-427, mit ihren sechs bis sieben identischen Nachbarn im Innern des beta-Fasses eine Einschnürung in der Röhre und das aktive Zentrum der Transportfunktion bildet. Diese Einschnürung bekam den Namen φ-Klemme (engl. φ-clamp), sie interagiert direkt mit dem zu transportierenden Protein.
Bryan A. Krantz schlug 2006 schließlich aufgrund der bisherigen Ergebnisse ein Modell für den Transportmechanismus der PA-63-Pore vor, bei dem der zu transportierende Faktor aufgrund des sauren Milieus nahezu entfaltet, das heißt als lineare Kette vorliegt, die nur mithilfe des pH-Gradienten und der Brownschen Molekularbewegung eine Netto-Bewegung aus dem Endosom ausführt. Die Vermutung dieses als „molekulare Ratsche“ bezeichneten Prinzips wurde bis heute nicht widerlegt.[11]
Letalfaktor (LF)
| Letalfaktor | ||
|---|---|---|
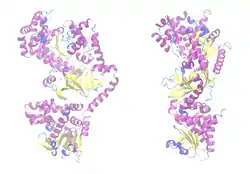
| ||
| Bändermodell des LF-Monomers, Zink als Kugel, nach PDB 1J7N | ||
|
Vorhandene Strukturdaten: s. UniProt | ||
| Masse/Länge Primärstruktur | 776 Aminosäuren | |
| Kofaktor | Zn++ | |
| Präkursor | prepro-LF (809 aa) | |
| Bezeichner | ||
| Gen-Name(n) | lef | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 3.4.24.83, Metallopeptidase | |
| MEROPS | M34.001 | |
| Reaktionsart | Hydrolyse von Peptidbindungen | |
| Substrat | alle MAP2K außer MAP2K5 | |
| Produkte | Abfallproteine | |
| Vorkommen | ||
| Homologie-Familie | Hovergen | |
Der Letalfaktor (LF) ist ein Enzym aus der Familie der neutralen Zink-Metallopeptidasen. Es katalysiert hochspezifisch die Hydrolyse von Peptidbindungen in bestimmten Enzymen der Wirtszelle, den MAP-Kinase-Kinasen (MAPKK, MAP2K, MEK). Es handelt sich um eine Endopeptidase.[7][18]
Beobachtet wurden bei der Verabreichung des Letaltoxins besonders zelltoxische Effekte auf alle Zellen. Diese zytotoxischen Effekte können auch durch ihre Ähnlichkeit mit der Wirkungsweise künstlicher MEK-Inhibitoren auf die Hemmung der MEK-Signalwege durch den Letalfaktor zurückgeführt werden.[7]
Die katalysierte Reaktion lautet:
- MEK + H2O Proteinbruchstücke
Die Enzymfunktion kommt nur bei Anwesenheit des Cofaktors Zink zustande.
Auch der Letalfaktor wird zunächst mit Signalsequenz synthetisiert, die nach dem Export via Sec-System abgeschnitten wird.
Ödemfaktor (EF)
| Ödemfaktor | ||
|---|---|---|
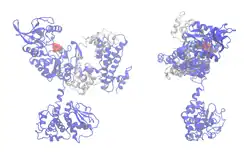
| ||
| Bändermodell des EF (blau) im Komplex mit Calmodulin (grau), Desoxy-ATP, Ca und Mg als Kalotten, nach PDB 1XVF. Die untere Domäne bindet an PA-63. | ||
| Masse/Länge Primärstruktur | 767 Aminosäuren | |
| Kofaktor | Ca++, Mg++, Calmodulin | |
| Präkursor | (800 aa) | |
| Bezeichner | ||
| Gen-Name(n) | cya | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 4.6.1.1, Lyase | |
| Reaktionsart | Ringschluss | |
| Substrat | ATP | |
| Produkte | 3',5'-cAMP + PPi | |
| Vorkommen | ||
| Homologie-Familie | Hovergen | |
Der Ödemfaktor (EF, auch genauer: Calmodulinsensitive Adenylylcyclase) von B. anthracis ist ein Enzym, das die Umwandlung von ATP in cAMP katalysiert. Es ist eine Adenylylcyclase der Klasse II. Die ungehemmte Produktion des unspezifischen Second Messengers cAMP löst, je nach Zelltyp, weitere Signale aus, deren Gesamtwirkung schwer zu überblicken ist. Jedoch wurden in letzter Zeit Hinweise gefunden, dass EF für die Schädigung des Immunsystems und der Blutgefäße verantwortlich ist, und damit zur Tödlichkeit der Infektion insgesamt beiträgt. EF allein wirkt nur in hohen Dosen tödlich auf die Zelle oder den gesamten Organismus.[7][19][20]
Das katalysierte Gleichgewicht lautet:
- ATP cAMP + PPi

Für ein funktionierendes Enzym ist dessen Komplexierung mit je einem Calcium- und Magnesiumion, sowie einem Molekül Calmodulin notwendig.
Auch der Ödemfaktor wird zunächst mit Signalsequenz synthetisiert, die nach dem Export via Sec-System abgeschnitten wird.
Evolution

Die Evolution des Milzbrandtoxins ist die Summe der Evolution seiner Protein-Untereinheiten. Obwohl das Pathogenitätsoperon vermutlich als Ganzes (so genannte Pathogenitätsinsel) auf das Plasmid pOX1 transponiert wurde, liegt dieses Ereignis weiter zurück als die Differenzierung der Gattungen Bacillus und Clostridium von ihrem gemeinsamen Vorgänger. Der Grund ist, dass mehrere Arten dieser Gattungen Proteine produzieren, die zu den Untereinheiten PA, LF und EF des Milzbrandtoxins homolog sind. Zusätzlich sind auch LF und EF zueinander homolog, so dass als gemeinsamer Urahn ein Enzym+PA-Doppeltoxin in Frage kommt. Daraus ergab sich die Benennung der Gruppe als A/B-Toxine.
Strukturbiologie-Datenbanken haben Aminosäure-Muster entwickelt, die auf alle so verwandten Proteine passen. Damit ist es nun möglich, direkt nach einer Genomsequenzierung entsprechende Gene sofort zu erkennen und zu markieren (Genannotation). Mit solchen Mustern und Hilfsmitteln wie dem BLAST-Algorithmus zeigt man leicht, dass die A-Untereinheiten (die Enzyme) stärker konserviert sind als das jeweilige PA, da sie einander ähnlicher sind.[21][22][23][24][25]
Schutzmöglichkeiten
Die Infektion mit Milzbrand ist mit Antibiotika behandelbar. Dies reicht jedoch oft nicht aus, Schädigungen durch das ausgeschüttete Toxin zu vermeiden. Zum Schutz der Nutztiere und des Menschen vor den Auswirkungen einer Infektion mit Milzbrand haben sich daher spezielle Impfstoffe und Gegenmittel etabliert. Auch ist die Empfindlichkeit auf das Toxin nicht bei allen Menschen gleich.
Die vom Ministerium für Innere Sicherheit der Vereinigten Staaten in großem Stil finanzierten Forschungsprojekte haben zu einer Reihe an Produkten geführt, die dem Land im Strategic National Stockpile zur Verfügung stehen. Trotz der Seltenheit möglicher Anwendungen liegen genug Daten vor, um die Effizienz maßgeschneiderter humaner monoklonaler Antikörper gegen die Milzbrandtoxin-Untereinheiten zu belegen. Sie können nun in den Fällen eingesetzt werden, die nicht mehr auf Antibiotika ansprechen.
Impfung
Die von Pasteur für Nutzvieh entwickelte Impfmethode bestand aus zwei Inokulationen im Abstand von zwei Wochen: Zellen aus B. anthracis-Kulturen, die unterschiedlich lange bei 42 bis 43 Grad attenuiert wurden. Eine solche Behandlung entfernt aus fast allen Zellen das Plasmid pOX1 und man vermutet, dass die folgende subklinische Infektion zum Erfolg der Impfung führt, da pOX1-freie Zellen keine Wirkung haben. Der Pasteur-Impfstoff wurde in den 1930er Jahren durch Carbozoo ersetzt, einen Impfstoff, bestehend aus Bakteriensporen in einer zehnprozentigen Saponin-Lösung. Daraus entwickelte sich der auch heute noch eingesetzte Sterne-Impfstoff aus Sporen des Stamms 34F2 in einer 0,5%igen Saponinlösung.[13]
Im Zuge des Kalten Krieges entwickelten die Großmächte in den 50er Jahren Impfstoffe für den Menschen, die heute kommerziell erhältlich sind: der russische Impfstoff basiert auf einer Abwandlung des Sterne-Stamms und wird mittels Skarifizierung angewandt. Seine Wirksamkeit ist unbekannt, im Gegensatz zur hohen Anzahl der Nebenwirkungen und Kontraindikationen. Die von den USA entwickelte Vakzine (Anthrax Vaccine Absorbed, AVA) wird von Emergent BioSolutions unter dem Handelsnamen BioThrax vertrieben; sie besteht aus in Fermentern produziertem PA auf einer Aluminiumhydroxid-Matrix. Das britische Produkt (Anthrax Vaccine Precipitated, AVP) unterscheidet sich davon nur geringfügig durch höheren LF- und EF-Anteil. AVA und AVP werden vom Militär prophylaktisch eingesetzt.[13][26][27]
Nach den Anschlägen vom 11. September 2001 und der folgenden Versendung von Milzbrandsporen stieg der Bedarf für einen bevölkerungsweit anwendbaren Impfstoff, den die militärischen Impfstoffe AVA und AVP nicht befriedigen konnten: insbesondere die Häufigkeit der notwendigen Anwendung (6 Injektionen über 18 Monate, danach einmal jährlich) ist für einen solchen Einsatz unakzeptabel. In der darauf einsetzenden Forschungstätigkeit wurden alle bekannten Methoden zur Impfstoffherstellung versucht. Wie bei vielen anderen Projekten zur Impfstoffherstellung gegen Bakterien blieben klinische Studien jedoch Mangelware. Deshalb und auch aufgrund der leichten Behandlung mit Antibiotika und humanen Antikörpern sind für neue Impfstoffe hohe Hürden gesetzt. Mit SparVax, einem Impfstoff aus rekombinantem, verändertem PA, hat die Firma PharmAthene ein Produkt entwickelt, das hochrein hergestellt und besser gelagert werden kann. In der nächsten Generation soll die Anzahl notwendiger Injektionen weiter reduziert werden.[26][28][29]
Seit 2013 ist durch das Paul-Ehrlich-Institut der azelluläre Impfstoff BioThrax®, dessen Wirkung auf die Antikörperinduktion gegen das Protektive Antigen beruht, zugelassen. Dadurch ist die aktive Immunisierung von Milzbranderregern exponierten Personen, wie Tierärzte und Abdecker möglich.[30]
Die Impfung von Tieren zum Schutz vor Milzbrand ist in Deutschland verboten. Impfstoffe für den Menschen sind in Deutschland nicht frei verfügbar und nicht zugelassen.
Gegenmittel
Humane monoklonale Antikörper (humAB) gegen die PA-Untereinheit im Milzbrandtoxin sind die Grundlage mehrerer Produkte, die für den Einsatz als Antitoxin entwickelt wurden. Sie können selbst noch Tage nach einer Infektion mit Milzbrandsporen die tödlichen Effekte des Toxins neutralisieren. Weitere niedermolekulare Verbindungen mit ähnlichen Wirkungen wurden entdeckt, es liegen jedoch noch keine klinischen Erfahrungen über sie vor.[31]
Der von emergent BioSolutions entwickelte humAB Thravixa befindet sich in Phase 1 der Zulassung. PharmAthene und Medarex sind mit ihrem als Orphan-Arzneimittel zugelassenen humAB Valortim bereits auf dem Markt und seine Weiterentwicklung wurde gefördert. Der high-affinity-humAB Anthim von Elusys, der ebenso von der FDA Orphan-Status erhielt, brachte dieser Firma bereits einen 140-Millionen-Dollar-Vertrag ein. Es ist davon auszugehen, dass nicht nur die genannten Firmen weitere Milzbrand-Antitoxine in der Pipeline haben.[32]
Bei der Suche nach niedermolekularen Verbindungen, die als Milzbrand-Antitoxin eingesetzt werden können, ergeben sich naturgemäß drei Hauptansätze: die Hemmung der PA-Transportfunktion und jeweils die Hemmung der LF- und EF-Enzymfunktionen. Die Hemmung der Transportfunktion kann außerdem unterschiedliche Ansätze haben: die Präsentation eines Rezeptors ohne Endozytosefähigkeit, Rezeptorantagonismus, sowie mehrere Möglichkeiten, die Ausbildung der Pore und ihre Funktion zu hemmen. Gefunden wurden daher bereits eine Vielzahl von Substanzen. Als Arzneistoffe mit ursprünglich anderer Indikation sind darunter Statine, Neomycin B und Verapamil.[31][33][34]
Natürliche Genvariationen
Das International HapMap Project analysiert weltweit menschliche Gene und ihre Variationen; dabei werden auch Zelllinien erfasst. Im Jahr 2011 konnten Martchenko und Mitarbeiter anhand von Lymphoblasten aus 234 Personen nachweisen, dass die Sensitivität dieser Zellen gegenüber PA-63 in einem weiten Bereich variierte und dass diese Sensibilität mit der Menge der produzierten mRNA korreliert, die für den Milzbrandtoxinrezeptor 2 (CMG-2) codiert. Die Untersuchung zeigte, dass die CMG-2-Expression bei verschiedenen Personen über vier Größenordnungen schwankt und Zellen dreier Europäer sogar unempfindlich auf Milzbrandtoxin sind. Die Vererbbarkeit der relativen Unempfindlichkeit konnte nachgewiesen werden; die Verteilung der Werte deutet auf Polygenie dieses Phänotyps.[35]
Einzelnachweise
- David P. Clark, Nanette J. Pazdernik: Molekulare Biotechnologie: Grundlagen und Anwendungen. Springer, 2009, ISBN 3-8274-2128-4, S. 563 ff.
- InterPro: IPR014781 Anthrax toxin, lethal/endema factor, N-/C-terminal (englisch)
- H. Katayama, B. E. Janowiak u. a.: GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. In: Nature structural & molecular biology. Band 15, Nummer 7, Juli 2008, S. 754–760, doi:10.1038/nsmb.1442. PMID 18568038. PMC 2504863 (freier Volltext).
- R. Koch: Untersuchungen über Bakterien: V. Die Ätiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus anthracis. (PDF; 11,1 MB). In: Beitrage zur Biologie der Pflanzen. 2 (2), 1876, S. 277–310. Cohns
- L. Pasteur, Chamberland und Roux: De l'attenuation des virus et de leur retour à la virulence. In: Comptes rendus. 92, 1881, S. 492
- P. Mikesell, B.E. Ivins u. a.: Plasmids, Pasteur, and Anthrax. (PDF; 199 kB) In: ASM News. 49, 1983, S. 320
- M. Moayeri, S. H. Leppla: Cellular and systemic effects of anthrax lethal toxin and edema toxin. In: Molecular Aspects of Medicine. Band 30, Nummer 6, Dezember 2009, S. 439–455. doi:10.1016/j.mam.2009.07.003. PMID 19638283. PMC 2784088 (freier Volltext). (Review).
- H. Smith, J. Keppie: Observations on experimental anthrax; demonstration of a specific lethal factor produced in vivo by Bacillus anthracis. In: Nature. Band 173, Nummer 4410, Mai 1954, S. 869–870. PMID 13165673.
- F. A. Beall, M. J. Taylor, C. B. Thorne: Rapid lethal effect in rats of a third component found upon fractionating the toxin of Bacillus anthracis. In: Journal of bacteriology. Band 83, Juni 1962, S. 1274–1280. PMID 13866126. PMC 279445 (freier Volltext).
- G. van der Goot, J. A. Young: Receptors of anthrax toxin and cell entry. In: Molecular aspects of medicine. Band 30, Nummer 6, Dezember 2009, S. 406–412, doi:10.1016/j.mam.2009.08.007. PMID 19732789. PMC 2783407 (freier Volltext). (Review).
- R. J. Collier: Membrane translocation by anthrax toxin. In: Molecular Aspects of Medicine. Band 30, Nummer 6, Dezember 2009, S. 413–422, doi:10.1016/j.mam.2009.06.003. PMID 19563824. PMC 2783560 (freier Volltext). (Review).
- R. T. Okinaka, K. Cloud u. a.: Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. In: Journal of bacteriology. Band 181, Nummer 20, Oktober 1999, S. 6509–6515. PMID 10515943. PMC 103788 (freier Volltext).
- R. Bhatnagar, S. Batra: Anthrax toxin. In: Critical reviews in microbiology. Band 27, Nummer 3, 2001, S. 167–200, doi:10.1080/20014091096738. PMID 11596878. (Review).
- J. G. Bann: Anthrax toxin protective antigen–insights into molecular switching from prepore to pore. In: Protein science : a publication of the Protein Society. Band 21, Nummer 1, Januar 2012, S. 1–12. doi:10.1002/pro.752. PMID 22095644.
- R. A. Pimental, K. A. Christensen u. a.: Anthrax toxin complexes: heptameric protective antigen can bind lethal factor and edema factor simultaneously. In: Biochemical and biophysical research communications. Band 322, Nummer 1, September 2004, S. 258–262, doi:10.1016/j.bbrc.2004.07.105. PMID 15313199.
- T. L. Nguyen: Three-dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. In: Journal of biomolecular structure & dynamics. Band 22, Nummer 3, Dezember 2004, S. 253–265. PMID 15473701.
- UniProt P09616
- A. D. Pannifer, T. Y. Wong u. a.: Crystal structure of the anthrax lethal factor. In: Nature. Band 414, Nummer 6860, November 2001, S. 229–233, doi:10.1038/n35101998. PMID 11700563.
- F. J. Maldonado-Arocho, J. A. Fulcher u. a.: Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. In: Molecular microbiology. Band 61, Nummer 2, Juli 2006, S. 324–337, doi:10.1111/j.1365-2958.2006.05232.x. PMID 16856939.
- W. J. Tang, Q. Guo: The adenylyl cyclase activity of anthrax edema factor. In: Molecular aspects of medicine. Band 30, Nummer 6, Dezember 2009, S. 423–430, doi:10.1016/j.mam.2009.06.001. PMID 19560485. PMC 2783455 (freier Volltext). (Review).
- S. Perelle, M. Gibert u. a.: Characterization of Clostridium perfringens iota-toxin genes and expression in Escherichia coli. In: Infection and Immunity. Band 61, Nummer 12, Dezember 1993, S. 5147–5156. PMID 8225592. PMC 281295 (freier Volltext).
- K. Kimura, T. Kubota u. a.: The gene for component-II of botulinum C2 toxin. In: Veterinary microbiology. Band 62, Nummer 1, April 1998, S. 27–34. PMID 9659689.
- S. Stubbs, M. Rupnik u. a.: Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. In: FEMS microbiology letters. Band 186, Nummer 2, Mai 2000, S. 307–312. PMID 10802189.
- InterPro: IPR003541 Anthrax toxin, lethal/endema factor (englisch)
- InterPro: IPR003896 Bacterial exotoxin B (englisch)
- R. J. Cybulski, P. Sanz, A. D. O'Brien: Anthrax vaccination strategies. In: Molecular aspects of medicine. Band 30, Nummer 6, Dezember 2009, S. 490–502, doi:10.1016/j.mam.2009.08.006. PMID 19729034. PMC 2783700 (freier Volltext). (Review).
- Webpräsenz von emergent
- T. Chitlaru, Z. Altboum u. a.: Progress and novel strategies in vaccine development and treatment of anthrax. In: Immunological Reviews. Band 239, Nummer 1, Januar 2011, S. 221–236, doi:10.1111/j.1600-065X.2010.00969.x. PMID 21198675. (Review).
- Webpräsenz von PharmAthene
- pei.de (Seite nicht mehr abrufbar, festgestellt im Mai 2019. Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.
- Y. Li, K. Sherer u. a.: New insights into the pathogenesis and treatment of anthrax toxin-induced shock. In: Expert opinion on biological therapy. Band 7, Nummer 6, Juni 2007, S. 843–854, doi:10.1517/14712598.7.6.843. PMID 17555370. (Review).
- K. Desai: Bioterrorism: Anthrax and Biotech Companies. In: seekingalpha.com, Hrsg.: Seeking Alpha. 17. Jun 2008, abgerufen am: 20. Feb 2012.
- M. Forino, S. Johnson u. a.: Efficient synthetic inhibitors of anthrax lethal factor. In: Proceedings of the National Academy of Sciences. Band 102, Nummer 27, Juli 2005, S. 9499–9504, doi:10.1073/pnas.0502733102. PMID 15983377. PMC 1160517 (freier Volltext).
- A. M. deCathelineau, G. M. Bokoch: Inactivation of rho GTPases by statins attenuates anthrax lethal toxin activity. In: Infection and immunity. Band 77, Nummer 1, Januar 2009, S. 348–359, doi:10.1128/IAI.01005-08. PMID 18936176. PMC 2612271 (freier Volltext).
- M. Martchenko, S. I. Candille u. a.: Human genetic variation altering anthrax toxin sensitivity. In: Proceedings of the National Academy of Sciences. Band 109, Nummer 8, Februar 2012, S. 2972–2977, doi:10.1073/pnas.1121006109. PMID 22315420. PMC 3286947 (freier Volltext).
Weblinks
- D. Goodsell: PDB Molecule of the Month 101: Anthrax Toxin. doi:10.2210/rcsb_pdb/mom_2002_4