Astrozytom, IDH-mutiert
Das Astrozytom, IDH-mutiert (kurz Astrozytom) ist ein diffus wachsender Hirntumor, dessen Zellen Astrozyten ähnlich sehen. Diese Gliazellen bilden das Stützgewebe des Zentralnervensystems, Astrozytome werden deshalb den Gliomen zugeordnet.
| Klassifikation nach ICD-10 | |
|---|---|
| D33 | Gutartige Neubildung des Gehirns und Zentralnervensystems |
| D43 | Neubildung unsicheren oder unbekannten Verhaltens des Gehirns und des Zentralnervensystems |
| C71 | Bösartige Neubildung des Gehirns |
| C72 | Bösartige Neubildung des Rückenmarkes, der Hirnnerven und anderer Teile des Zentralnervensystems |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9400/3 | Astrozytom, IDH-mutiert, Grad 2 |
| 9401/3 | Astrozytom, IDH-mutiert, Grad 3 |
| 9445/3 | Astrozytom, IDH-mutiert, Grad 4 |
| ICD-O-3, zweite Revision (2019) | |

In der WHO-Klassifikation der Tumoren des zentralen Nervensystems wird das IDH-mutierte Astrozytom anhand molekulargenetischer und histologischer Eigenschaften definiert. Insbesondere in Folge von Fortschritten der Molekulargenetik vereinigt der Tumortyp die früheren Entitäten IDH-mutiertes Glioblastom, anaplastisches Astrozytom und diffuses Astrozytom, Letzteres mit den Varianten gemistozytisches, fibrilläres und protoplasmatisches Astrozytom. Abhängig vom Verhalten des Tumors und seiner Prognose werden Astrozytomen die ZNS WHO Grade 2 bis 4 zugeordnet.
Die Therapie erfolgt mittels Operation, Bestrahlung und Chemotherapeutika.
Epidemiologie
Durch die veränderte Klassifikation liegen mit Stand 2023 noch keine genauen Daten zur Häufigkeit vor. Nach alter Klassifikation traten diffuse (Grad 2) und anaplastische (Grad 3) Astrozytome mit einer Inzidenz von 0,5 bzw. 0,4 Fällen pro 100.000 Menschen pro Jahr auf. Das mediane Alter der Patientinnen und Patienten mit diesem Tumor liegt bei 35 Jahren, Männer sind etwas häufiger betroffen als Frauen. Niedriggradige Subtypen treten eher bei jüngeren Menschen auf, höhergradige eher bei älteren.[1][2]
Krankheitsentstehung und Risikofaktoren
Welche Zellart oder Zellarten Ursprung IDH-mutierter Astrozytome sein können, ist, wie bei anderen Gliomen, nicht abschließend geklärt. In den Tumoren finden sich Zellen mit astrozytärer, oligodendrozytärer und neuroblastärer Differenzierung.[1]
Die meisten Fälle sind sporadisch. Familiäre Häufungen treten bei bestimmten einzelnen Genmutationen und manchen Syndromen genetischer Instabilität (z. B. Li-Fraumeni-Syndrom) auf.[1]
Lokalisation
Astrozytome finden sich vorwiegend im Frontal- und Temporallappen des Großhirns. Die Tumoren entwickeln sich im Bereich der weißen Substanz (Nervenzellfaserbündel) der Hemisphären (Hirnhälften) und liegen meist „unterhalb“ der Hirnrinde (subkortikal). Sie können aber auch in allen anderen Abschnitten des Gehirns und des Rückenmarks auftreten.
Wie die meisten anderen Gliome metastasieren Astrozytome nur selten.[1]
Symptome
Bei etwa zwei Drittel der Patientinnen und Patienten ist das erste klinische Symptom ein epileptischer Anfall.[3] Der Mechanismus der Symptome ist die Infiltration und Zerstörung benachbarter Neurone. Durch einen Verdrängungsdruck kommt es zum „Hirndruck“. Das häufigste gemeinsame Zeichen dieser Mechanismen ist ein Papillenödem (Vorwölbung der Papille, der Austrittsstelle des Sehnerven in der Netzhaut, ohne Minderung der Sehkraft). Kopfschmerzen, Lethargie und Persönlichkeitsveränderungen sind ebenfalls häufige Zeichen eines beginnenden Hirndruckes. Fokale neurologische Zeichen (Lähmung, Störung der Hirnnervenfunktion, Kopfschmerzen) gehen der Diagnosestellung oft Jahre voraus.
Technische Untersuchungsbefunde
Bildgebung

Die Bildgebung erfolgt in der Regel mittels Magnetresonanztomographie. Eine Kontrastmittelaufnahme ist bei Grad-2-Astrozytomen selten (20 %), bei höhergradigen Subtypen jedoch häufig. Verkalkungen können vorliegen. Insbesondere höhergradige Tumoren können auch ein umliegendes Ödem zeigen.[2][4][5]
Ein hochsensitives Merkmal für Astrozytome der Grade 2 und 3 ist ein T2/FLAIR-Mismatch, also ein Darstellungsunterschied zwischen zwei MRT-Sequenzen. In der T2-Sequenz zeigt sich der gesamte Tumorbereich hyperintens, also aufgehellt, während in der FLAIR-Sequenz nur der Randsaum hyperintens ist. In der T1-Sequenz ist der Tumor hypointens.[1][2][3][6]
Darstellung von metabolischen Eigenschaften ist mittels Magnetresonanzspektroskopie möglich.[2][5]
Astrozytome können auch als Zufallsbefund bei computertomographischen Untersuchungen finden. Grad 2 zeigt unscharfe Hypodensitäten, manchmal auch Verkalkungen oder zystische Formationen. Höhere Grade präsentieren auch Ödeme oder Kontrastmittelanreicherung. Insbesondere Astrozytome Grad 4 können sich ähnlich wie Glioblastome mit großen, zentralen Nekrosen mit umgebender Kontrastmittelanreicherung darstellen.[1]

Im PET-Scan des Glucose-Stoffwechsels (FDG-PET) stellt sich das Astrozytom hypometabolisch dar („kalter Knoten“, das heißt, es ist Gewebe mit vermindertem Stoff- und Energieumsatz). Entdifferenzierungen innerhalb des Tumors führen gelegentlich zu malignen Zwischenstufen, die dann im PET-Bild als „hot spots“ (Gewebe mit erhöhtem Stoff- und Energieumsatz) innerhalb des „kalten Knotens“ erscheinen können.
In der Angiographie zeigen Astrozytome typischerweise keine pathologische Blutgefäßarchitektur (Vaskularisierung).
Liquor
Der Liquorbefund ist in der Regel unauffällig.
Pathologie
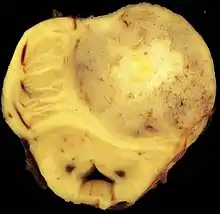
IDH-mutierte Astrozytome wachsen expansiv und diffus infiltrierend, die Grenze zwischen grauer und weißer Substanz kann verwaschen sein. Blutgefäße werden verdrängt, die Hirnhäute können infiltriert sein und der Tumor kann zum Beispiel eine Gewebebrücke durch die Sylvische Fissur bilden. Eine Liquoraussaat von Tumorzellen ist selten. Selten gibt es degenerative Veränderungen innerhalb von Mikrozysten mit Verkalkungen. Bei Grad 4 kann es zu Nekrosen und Einblutungen kommen.
Histologie
Astrozytome bestehen aus Gliazellen, welche diffus das Gewebe durchwachsen. Dabei reicht das Spektrum von nur geringgradig veränderten Gewebeproben bis zu hochmalignen, hyperzellulären Tumoren. Insbesondere bei Astrozytomen Grad 2 lassen sich verschiedene Differenzierungstypen ausmachen, die früher als eigenständige Varianten des diffusen Astrozytoms geführt wurden. Die am häufigsten vorkommenden fibrilläre Differenzierung zeigt eine eher lockere Durchsetzung des Gewebes mit Gliafasern. Häufig finden sich Anteile mit gemistozytärer Differenzierung, dabei handelt es sich um Tumorzellen mit großen, eosinophilem Zytoplasmata und teils mehreren exzentrisch gelegenen Kernen.[1][7]
Bei den Graden 3 und 4 finden sich Anaplasien. Ausschließlich bei Grad 4 kann es auch zu einem raschen Wachstum kleiner Blutgefäße, der mikrovaskulären Proliferation, kommen. Atypische Zellkerne kommen bei allen Graden vor, sind jedoch nicht für die Gradierung entscheidend.[1][7]
Genetik
Beim IDH-mutierten Astrozytom liegt eine Gain-of-function-Mutation der Isocitrat-Dehydrogenase (IDH) vor. In 90 % der Fälle handelt es sich um die R132H-Mutation von IDH1, bei der eine einzige Aminosäure an Position 132 – Arginin (R) gegen Histidin (H) – ausgetauscht ist. In je 5 % der Fälle liegt eine atypische Mutationen von IDH1 oder IDH2 vor, diese sind vor allem bei jüngeren Menschen häufiger. Diese Mutationen beeinträchtigen die Umwandlung von Isocitrat in alpha-Ketoglutarat, was für die zelluläre Energiebereitstellung notwendig ist, und reduzieren es stattdessen direkt zu 2-Hydroxyglutarat, dessen erhöhter Spiegel epigenetische Veränderungen, vor allem Hypermethylierungen, verursacht.[8] Dieser Zustand wird als glioma-associated CpG island methylator phenotype (G-CIMP) bezeichnet und geht mit veränderter Expression diverser Regulatoren des Zellwachstums, sogenannter Onko- und Tumorsuppressorgene, einher. Eine hohe Gesamtzahl an Mutationen, viele Kopienzahlveränderungen und ein geringer Grad der DNA-Methylierung sind mit schlechteren Prognosen assoziiert.[1][9][10][11]
Eine mögliche Erklärung für die bevorzugte Lage im Frontallappen ist die dortige Expression des Gens GLUD2, welches für die Glutamatdehydrogenase (GDH) kodiert. Die GDH katalysiert ebenfalls die Synthese von alpha-Ketoglutarat und kann so die negativen Auswirkungen der IDH-Mutation auf die mitochondriale Energiebereitstellung teilkompensieren.[1]
Etwa 70 bis 90 % der IDH-mutierten Astrozytome haben eine Loss-of-Function-Mutation in den Tumorsuppressorgenen TP53 und ATRX. Der Verlust des ATRX-Proteins ermöglicht Krebszellen die Telomerase-unabhängige Verlängerung der Telomere und damit mehr Zellteilungen. TP53 kodiert für das Protein p53, einen wichtigen Initiator des programmierten Zelltods. Homozygote Deletionen, also der Verlust beider Kopien, von CDKN2A- und CDKNA2B sind mit schlechterer Prognose assoziiert, diese Tumoren werden als Grad 4 eingestuft.[5][11][12][13]
Eine 1p/19q-Kodeletion schließt TP53- und ATRX-Mutationen oder -Verluste weitestgehend aus, IDH-mutierte Gliome mit dieser Kodeletion werden als Oligodendrogliom, IDH-mutiert und 1p/19q-kodeletiert klassifiziert. Anhand dieses Kriteriums können auch fast alle ehemals als Oligoastrozytome (auch Mischgliome) bezeichnete Tumoren entweder als Astrozytom oder Oligodendrogliom identifiziert werden.[2][9]
Epigenetik
Die O6-Methylguanin-DNA-Methyltransferase repariert alkylierte DNA und schützt den Körper damit vor der Entstehung von Tumoren. In etwa 85 % der Astrozytome ist das Enzym durch die Hypermethylierung des MGMT-Promotors weitestgehend inaktiv.[5]
Immunhistochemie
Über immunhistochemische Verfahren können diverse Surrogatmarker für die Zellteilungsrate oder genetische und epigenetische Veränderungen dargestellt werden. Wie bei anderen Gliomen sind das saure Gliafaserprotein und Vimentin fast immer darstellbar und ermöglichen die Identifikation der diffusen Infiltration. Zudem können verschiedene IDH-Mutationen, ATRX- und TP53-Mutationen oder -Verluste, der Zellteilungsmarker Ki-67 und OLIG2 angefärbt werden. Bei Nachweis von ATRX- oder TP53-Mutationen kann auf den genetischen Test einer 1p/19q-Kodeletion zur Differentialdiagnostik eines Oligodendroglioms verzichtet werden. Der Verlust von p16 oder MTAP als Surrogat für eine homzygote CKDN2A/B-Deletion ist Gegenstand aktueller Forschung.[7][9][12]
Gradierung

Das Astrozytom hat nach der WHO-Klassifikation von 2021 drei Subtypen, welche anhand molekulargenetischer und histologischer Kriterien unterschieden werden. Nach der Version von 2016 gab es diverse eigenständige Tumorentitäten und -varianten, die in den Subtypen aufgegangen sind.[1][10]
Strikte Grenzwerte für Mitosenzahl, Ki-67-Index oder ähnlichem zur genauen Abgrenzung der Grade 2 und 3 liegen mit Stand 2023 nicht vor.[5][7]
- Astrozytom, IDH-mutiert, Grad 2
- Das Astrozytom Grad 2 zeigt histologisch keine Anaplasien, Nekrosen oder mikrovaskuläre Proliferation, das Gewebe kann jedoch etwas verdichtet sein und atypische Mitosen zeigen. Üblicherweise sind weniger als vier Prozent der Zellen Ki-67-positiv. Der Unterscheidung des hochdifferenzierten Gewebes von einer Astrogliose oder gesunden Astrozyten erfolgt mittels immunhistochemischen Nachweises der IDH-Mutation. In vorherigen Versionen der WHO-Klassifikation wurden diese Subtypen als diffuses Astrozytom mit den Varianten gemistozytisches, fibrilläres und protoplasmatisches Astrozytom bezeichnet.[1][2][12]
- Astrozytom, IDH-mutiert, Grad 3
- Das Astrozytom Grad 3 zeigt histologisch Anaplasien, verstärkte mitotische Aktivität (Ki-67 4–10 %), dabei auch atypische Teilungen und vielkernige Zellen. Diese Tumoren wurden früher meist als anaplastisches Astrozytom eingestuft.[1][2]
- Astrozytom, IDH-mutiert, Grad 4
- Nur das Astrozytom Grad 4 zeigt hohe mitotische Aktivität, Anaplasien, mikrovaskuläre Proliferation, Nekrosen und/oder homozygote CKDN2A/B-Deletionen. Hierbei handelt es sich häufig um früher als IDH-mutierte Glioblastome klassifizierte Tumoren, mit denen sie häufige TERT-Mutationen und/oder einen +7/-10-Genotyp gemeinsam haben. Sie entstehen meistens de novo, entwickeln sich also nicht, wie lange gedacht, aus niedriggradigeren Astrozytomen.[1][2][10]
Therapie
Die Therapie des IDH-mutierten Astrozytoms ist vom Allgemeinzustand der Patientinnen und Patienten, den Therapiezielen und Grad und Lokalisation des Tumors abhängig. Sie wird interdisziplinär im Rahmen eines Tumorboards zwischen der Neuroonkologie, Neuroradiologie, Neuropathologie, Neurochirurgie und Radioonkologie besprochen. Die Standardtherapie ist eine Kombination aus chirurgischer Resektion, Bestrahlung und Chemotherapie, ergänzt durch supportive Maßnahmen.[2][14]
Chirurgische Therapie

Die neurochirurgische Operation mit Verminderung der Hauptmasse des Tumors (Tumorreduktion) kann das Fortschreiten der Erkrankung verlangsamen, aber nicht dauerhaft verhindern, da praktisch immer einzelne Tumorzellen das gesunde Gehirngewebe schon infiltrativ durchwandert haben und deswegen eine vollständige Tumorentfernung nicht möglich ist. Eine möglichst frühe und weitreichende Resektion ist ein wichtiger prognostischer Faktor. Auf eine Biopsie sollte nur Ausnahmefällen verzichtet werden.[2]
Bei Astrozytomen vom Grad 2 kann insbesondere bei jüngeren Patientinnen und Patienten nach einer Resektion zunächst auf eine Bestrahlung und Chemotherapie verzichtet werden, falls der Tumor besonders klein und größenstabil ist und keine Symptome verursacht.[2][4]
Radiotherapie
Eine Bestrahlung des Tumors und Teile der Umgebung verlängert das progressionsfreie Überleben deutlich. Insbesondere bei niedriggradigen Astrozytomen mit langsamerem Wachstum muss besondere Rücksicht auf umliegendes gesundes Gewebe genommen werden, um die Nebenwirkungen der Therapie zu minimieren.[2]
Eine Bestrahlung ist mit höheren Raten an homozygoter CDKN2A-Deletion bei Rezidiven assoziiert.[12]
Chemotherapie
Die etabliertesten Zytostatika in der Behandlung des Glioblastoms sind Alkylanzien, welche die DNA-Replikation stören und so insbesondere das Wachstum sich schnell vermehrender Tumorzellen hemmen.
Astrozytome von Grad 2 werden mit einer kombinierten Chemotherapie aus Procarbazin, CCNU (Lomustin) und dem Mitosehemmer Vincristin (PCV-Schema) behandelt. Alternativ kann das Alkylanz Temozolomid zum Einsatz kommen, welches bei den Graden 3 und 4 Standard ist. Bei den Graden 2 und 3 erfolgt die Chemotherapie erhaltend nach einer Bestrahlung, bei Grad 4 analog zum Glioblastom auch währenddessen.[2]
Therapie bei Progression und Rezidiven
Grundsätzlich können alle Therapieformen auch in der Therapie von Rezidiven und progressiven Tumoren angewandt werden, wobei klinische Daten zur Effektivität häufig noch nicht eindeutig sind. Ein Einschluss in klinische Studien sollte erwogen werden.[2]
Supportivtherapie
Ein perifokales Hirnödem kann mit Corticosteroiden reduziert werden. Bei epileptischen Anfällen sollte eine Therapie mit Antiepileptika eingeleitet werden.[2][14]
Psychosoziale Versorgung
Astrozytompatientinnen und -patienten sollte psychologische Beratung und Therapie angeboten werden. Ebenso können sie nach Bedarf logopädische, ergo- und physiotherapeutische Behandlungen und sozialarbeiterische Unterstützung erhalten.[2][14]
Forschung
Klassifikation
Vor allem anhand von molekulargenetischen Markern werden, mit Stand 2023, für manche IDH-mutierte Astrozytome eine Klassifikation als eigener Tumortyp vorgeschlagen.
Das infratentorielle Astrozytom, IDH-mutiert ist sehr selten, bis 2023 wurden weltweit 40 Fälle beschrieben. Namensgebend ist die Lokalisation unterhalb des Tentoriums im Hirnstamm oder Kleinhirn. Es weist meist atypische IDH-Mutationen auf, zeigt geringeren ATRX-Verlust und/oder MGMT-Poromotor-Methylierung und weitere epigenetische Unterschiede. Teilweise liegt auch eine Histon-H3-Mutation vor, welche sonst charakteristisch für diffuse Mittelliniengliome (DMG) ist. Die Prognose ist besser als bei DMGs, jedoch schlechter als bei sonstigen IDH-mutierten Astrozytomen.[15][16][17][18]
Ein primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA, deutsch „primäres Mismatch-Reparatur-defizientes IDH-mutiertes Astrozytom“) tritt vor allem bei Kindern im Rahmen eines Mismatch-Reparatur-Defizienz-Syndroms (häufig MSH6-Mutation) oder Lynch-Syndrom auf. PMMRDIAs weisen eine hohe Mutationslast und eine schlechte Prognose auf.[15][16][19]
Therapien
Forschungsansätze für gezielte Therapien zielen vielfach auf mutiertes IDH und seine Auswirkungen. IDH-Hemmer wie Ivosidenib und Vorasidenib sind bereits für die IDH-mutierte akute myeloische Leukämie zugelassen und werden in ihrer Wirkung auf Gliome erforscht. 2023 finden sich Immuntherapien mit Checkpoint-Inhibitoren wie Pembrolizumab oder Avelumab und Peptidimpfstoffe gegen die IDH1-R132H-Mutation in frühen klinischen Studien.[8][12]
CDK4- und CDK6-Inhibitoren wie Palbociclib sind für die Behandlung mancher Brustkrebsvarianten zugelassen. Ihre Effektivität wird bei Gliomen mit der prognostisch ungünstigen homozygoten CDKN2A/B-Deletion erforscht.[12]
Die Kombination des Anti-VEGF-Antikörpers Bevacizumab mit Temozolomid bringt gegenüber einer alleinigen Temozolomidtherapie keinen Überlebensvorteil.[2]
Prognose
IDH-mutierte Astrozytome sind nicht heilbar, die mittlere Überlebenszeit hängt vom Grad des Tumors und weiteren Einflüssen ab. Günstige prognostische Faktoren sind unter anderem junges Alter und eine möglichst restlose Resektion. Die mediane Überlebenszeit beträgt bei Grad 2 über zehn Jahre, bei Grad 3 fünf bis zehn Jahre und bei Grad 4 etwa drei Jahre.[1]
Literatur
- S2k-Leitlinie Gliome der Deutsche Gesellschaft für Neurologie. In: AWMF online (Stand 01.02.2021, inhaltlich überprüft am 31.10.2023)
Einzelnachweise
- WHO Classification of Tumours Editorial Board (Hrsg.): Central Nervous System Tumours (= World Health Organization Classification of Tumours). 5th ed Auflage. International Agency for Research on Cancer, Lyon 2021, ISBN 978-92-832-4508-7 (englisch).
- S2k-Leitlinie Gliome der Deutsche Gesellschaft für Neurologie. In: AWMF online (Stand 01.02.2021, inhaltlich überprüft am 31.10.2023)
- Allan H. Ropper, Martin A. Samuels, Joshua P. Klein, Sashank Prasad: Adams and Victor's principles of neurology. Twelfth edition Auflage. McGraw Hill, New York Chicago San Francisco Athens London 2023, ISBN 978-1-264-26452-0, S. 651 ff.
- Antonio Dono, Leomar Y. Ballester, Ditte Primdahl et al.: IDH-Mutant Low-grade Glioma: Advances in Molecular Diagnosis, Management, and Future Directions. In: Current Oncology Reports. Band 23, Nr. 2, Februar 2021, ISSN 1523-3790, doi:10.1007/s11912-020-01006-6 (springer.com [abgerufen am 16. Dezember 2023]).
- Julie J Miller, L Nicolas Gonzalez Castro, Samuel McBrayer et al.: Isocitrate dehydrogenase (IDH) mutant gliomas: A Society for Neuro-Oncology (SNO) consensus review on diagnosis, management, and future directions. In: Neuro-Oncology. Band 25, Nr. 1, 5. Januar 2023, ISSN 1522-8517, S. 4–25, doi:10.1093/neuonc/noac207, PMID 36239925, PMC 9825337 (freier Volltext).
- Ziqin Han, Qiuying Chen, Lu Zhang et al.: Radiogenomic association between the T2-FLAIR mismatch sign and IDH mutation status in adult patients with lower-grade gliomas: an updated systematic review and meta-analysis. In: European Radiology. Band 32, Nr. 8, August 2022, ISSN 1432-1084, S. 5339–5352, doi:10.1007/s00330-022-08607-8.
- B.K. Kleinschmidt-DeMasters, Melike Pekmezci, Fausto Rodriguez, Tarik Tihan: Diagnostic Pathology: Neuropathology. 3. Auflage. Elsevier, 2022, ISBN 978-0-323-71331-3, S. 4 ff. (englisch).
- Sue Han, Yang Liu, Sabrina J. Cai et al.: IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. In: British Journal of Cancer. Band 122, Nr. 11, 26. Mai 2020, ISSN 0007-0920, S. 1580–1589, doi:10.1038/s41416-020-0814-x, PMID 32291392, PMC 7250901 (freier Volltext).
- Serena Ammendola, Giuseppe Broggi, Valeria Barresi: IDH-mutant diffuse gliomas: Tips and tricks in the era of genomic tumor classification. In: Histology and Histopathology. Band 38, Nr. 07, 2023, ISSN 0213-3911, S. 739–753, doi:10.14670/HH-18-582.
- Patrick Y Wen, Roger J Packer: The 2021 WHO Classification of Tumors of the Central Nervous System: clinical implications. In: Neuro-Oncology. Band 23, Nr. 8, 2. August 2021, ISSN 1522-8517, S. 1215–1217, doi:10.1093/neuonc/noab120, PMID 34185090, PMC 8328017 (freier Volltext).
- C. Mircea S. Tesileanu, Wies R. Vallentgoed et al.: Molecular markers related to patient outcome in patients with IDH-mutant astrocytomas grade 2 to 4: A systematic review. In: European Journal of Cancer. Band 175, November 2022, S. 214–223, doi:10.1016/j.ejca.2022.08.016 (elsevier.com [abgerufen am 16. Dezember 2023]).
- Simon Gritsch, Tracy T. Batchelor, L. Nicolas Gonzalez Castro: Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system. In: Cancer. Band 128, Nr. 1, Januar 2022, ISSN 0008-543X, S. 47–58, doi:10.1002/cncr.33918.
- David.E. Reuss: Updates on the WHO diagnosis of IDH-mutant glioma. In: Journal of Neuro-Oncology. Band 162, Nr. 3, Mai 2023, ISSN 0167-594X, S. 461–469, doi:10.1007/s11060-023-04250-5, PMID 36717507, PMC 10227121 (freier Volltext).
- Michael Weller, Martin van den Bent, Matthias Preusser et al.: EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. In: Nature Reviews Clinical Oncology. Band 18, Nr. 3, März 2021, ISSN 1759-4774, S. 170–186, doi:10.1038/s41571-020-00447-z, PMID 33293629, PMC 7904519 (freier Volltext).
- Julie J Miller, L Nicolas Gonzalez Castro, Samuel McBrayer et al.: Isocitrate dehydrogenase (IDH) mutant gliomas: A Society for Neuro-Oncology (SNO) consensus review on diagnosis, management, and future directions. In: Neuro-Oncology. Band 25, Nr. 1, 5. Januar 2023, ISSN 1522-8517, S. 4–25, doi:10.1093/neuonc/noac207, PMID 36239925, PMC 9825337 (freier Volltext).
- Serena Ammendola, Giuseppe Broggi, Valeria Barresi: IDH-mutant diffuse gliomas: Tips and tricks in the era of genomic tumor classification. In: Histology and Histopathology. Band 38, Nr. 07, 2023, ISSN 0213-3911, S. 739–753, doi:10.14670/HH-18-582.
- David.E. Reuss: Updates on the WHO diagnosis of IDH-mutant glioma. In: Journal of Neuro-Oncology. Band 162, Nr. 3, Mai 2023, ISSN 0167-594X, S. 461–469, doi:10.1007/s11060-023-04250-5, PMID 36717507, PMC 10227121 (freier Volltext).
- Rouzbeh Banan, Damian Stichel, Anja Bleck et al.: Infratentorial IDH-mutant astrocytoma is a distinct subtype. In: Acta Neuropathologica. Band 140, Nr. 4, Oktober 2020, ISSN 0001-6322, S. 569–581, doi:10.1007/s00401-020-02194-y.
- Abigail K. Suwala, Damian Stichel, Daniel Schrimpf et al.: Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. In: Acta Neuropathologica. Band 141, Nr. 1, Januar 2021, ISSN 0001-6322, S. 85–100, doi:10.1007/s00401-020-02243-6, PMID 33216206, PMC 7785563 (freier Volltext).