Allan-Herndon-Dudley-Syndrom
Das Allan-Herndon-Dudley-Syndrom (AHDS) ist eine seltene x-chromosmal vererbte Erkrankung des zentralen Nervensystems (X-chromosomale mentale Retardierung), die zu schweren geistigen Entwicklungsverzögerungen und Störungen der Motorik führt. Diese Erkrankung, die nur männliche Säuglinge betrifft, führt bereits vor der Geburt zu einer Störung der Entwicklung.
Das erstmals im Jahre 1944 von Wiliam Allan, Florence C. Dudley und C. Nash Herndon[1] beschriebene Syndrom ist Folge der gestörten Bildung zweier Schilddrüsenhormontransporter. Diese führt dazu, dass Schilddrüsenhormone von Zellen des Nervensystems, die darauf angewiesen sind, nicht aufgenommen werden.
Symptomatik
Betroffene Kinder fallen früh durch Muskelschwäche (Myasthenie) und eine unterentwickelte Muskulatur auf. Später können Gelenkdeformitäten und Kontrakturen auftreten, die zu einer zusätzlichen Beeinträchtigung der Beweglichkeit führen, ebenso wie Muskelkrämpfe und unwillkürliche Bewegungen der Arme und Beine. Die meisten Personen, die an AHDS leiden, sind daher nicht in der Lage, selbständig zu gehen. Betroffene leiden darüber hinaus an schweren geistigen Störungen, die so erheblich sind, dass sie meist nicht in der Lage sind, zu sprechen.
Ätiologie und Pathogenese
Nach Muskelverletzungen werden Stammzellen des Skelettmuskels aktiviert. Diese teilen sich und ersetzen geschädigte Muskelzellen – der Muskel heilt. Gesunde Muskelstammzellen bilden nach Verletzungen zwei Transporter für Schilddrüsenhormone verstärkt neu: MCT8 und OATP1C1. Ersteres ist beim AHDS durch eine Mutation des SCL16A2-Gens defekt. MCT8 vermittelt die Jodothyronin-Aufnahme in Muskel- und Nervenzellen. An sich nehmen Stammzellen beide Hormone auf und lassen den Muskel heilen. Schaltete man in gesunden Muskelstammzellen beide Transporter ab, heilten verletzte Muskelzellen nur langsam. Beide Transporter vermitteln auch im Gehirn die Aufnahme von Schilddrüsenhormonen über die Blut-Hirn-Schranke.[2]
Beim AHDS fehlt in den Muskeln und im Gehirn das aktive Schilddrüsenhormon T3. Laborchemisch liegen erhöhte T3-Spiegel (High-T3-Syndrom) vor, während die TSH- und FT4-Spiegel normal sind. Früher wurde diskutiert, ob dies Folge einer verminderten T3-Aufnahme in bestimmte Zelltypen ist oder von einer kompensatorischen Hyperdejodierung herrührt. Eine dritte Hypothese ging davon aus, dass es sich um die Folgen eines verminderten Austransports von Thyroxin aus Schilddrüsenzellen mit konsekutiver substratvermittelter Überaktivität intrathyreoidaler Dejodinasen handelt[3]. Diese Annahme wird durch die Ergebnisse von in-silico-Experimenten mit Computersimulationen unterstützt[4].
Erblichkeit
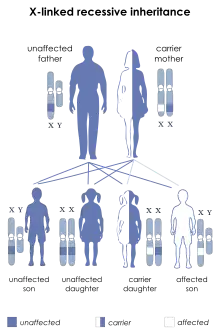
Die Erkrankung ist X-chromosomal-rezessiv vererbt. Frauen, die in jeder Zelle zwei X-Chromosomen haben, können die Erkrankung vererben, erkranken aber in der Regel selbst nicht, da die Wahrscheinlichkeit, dass beide X-Chromosomen ein mutiertes SLC16A2-Gen tragen, nur winzig ist. Männer haben nur ein X-Chromosom, so dass sich bei ihnen die Erkrankung im Falle einer Mutation manifestiert.
Bislang wurde noch nicht beobachtet, dass eine Frau an AHDS leidet. Neben der geringen Verbundwahrscheinlichkeit für SLC16A2-Mutationen in beiden X-Chromosomen dürfte auch dazu beitragen, dass erkrankte Männer nicht in der Lage sind, sich fortzupflanzen.
Therapie
Bis jetzt gibt es keine etablierte Therapie des AHDS, allerdings laufen derzeit klinische Studien. Aus theoretischen Erwägungen sollte TRIAC (Triiodothyroacetat, ein auch normalerweise im Körper gebildetes nicht klassisches Schilddrüsenhormon) von Nutzen sein, jedoch half dies in frühen klinischen Studien Kindern nicht merklich – evtl. weil die Therapie hierbei zu spät begann oder die Dosis nicht hinreichte. Im Jahre 2014 wurde allerdings ein Fall beschrieben, in dem eine Therapie mit TRIAC, die im Säuglingsalter begonnen wurde, die geistige Entwicklung und Mobilität merklich verbesserte.[5] Eine kürzlich veröffentlichte Studie konnte nun sowohl die Effektivität als auch die Sicherheit einer Therapie mit TRIAC demonstrieren.[6]
Weblinks
- Allan-Herndon-Dudley-Syndrom (AHDS), Eintrag bei orpha.net; abgerufen am 17. Januar 2015
- Eintrag 300523 bei OMIM
Einzelnachweise
- W. Allan, C. N. Herndon, F. C. Dudley. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. In: Am. J. Ment. Defic., 1944, 48, S. 325–334.
- Steffen Mayerl, Manuel Schmidt, Denica Doycheva, Veerle M. Darras, Sören S. Hüttner, Anita Boelen, Theo J. Visser, Christoph Kaether, Heike Heuer, Julia von Maltzahn: Thyroid Hormone Transporters MCT8 and OATP1C1 Control Skeletal Muscle Regeneration. In: Stem Cell Reports, 10, 2018, S. 1959, doi:10.1016/j.stemcr.2018.03.021.
- J Müller, H Heuer: Understanding the hypothalamus-pituitary-thyroid axis in mct8 deficiency. In: European Thyroid Journal. 1. Jahrgang, Nr. 2, Juli 2012, S. 72–9, doi:10.1159/000339474, PMID 24783000, PMC 3821472 (freier Volltext).
- TM Wolff, C Veil, JW Dietrich, MA Müller: Mathematical modeling and simulation of thyroid homeostasis: Implications for the Allan-Herndon-Dudley syndrome. In: Frontiers in Endocrinology. 13. Jahrgang, 2022, S. 882788, doi:10.3389/fendo.2022.882788, PMID 36568087, PMC 9772020 (freier Volltext).
- Ainhoa Iglesias, Olga Esther Alonso, Ana Gómez-Gila, Juan A. Campos-Cerveró, María Palomares, Eduvigis Contreras, Beatriz Morte, Maria Jesús Obregón, Juan Bernal, José C. Moreno: Triac Treatment Of An Infant With Allan-Herndon-Dudley Syndrome (Ahds): Effects On Iodothyronines In Serum And Cerebro-Spinal Fluid. (PDF; 363 kB) 38th Annual Meeting of the European Thyroid Association, Santiago de Compostela, Spain, September 10th, 2014.
- Stefan Groeneweg, Robin P Peeters u. a.: Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. In: The Lancet Diabetes & Endocrinology. 7, 2019, S. 695–706, PMID 31377265. doi:10.1016/S2213-8587(19)30155-X.