2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosylbromid
2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromid (Acetobromglucose) ist ein so genanntes Glycosylhalogenid[7] und als Glycosyldonor ein Standardreagenz für Glycosylierungsreaktionen in der Kohlenhydratchemie. Mit geeigneten Glycosylakzeptoren reagiert Acetobromglucose in Gegenwart von Silbersalzen nach der Koenigs-Knorr-Methode unter Ausbildung glycosidischer Bindungen zu Glucosiden, einer Untergruppe der Glycoside.[8]
| Strukturformel | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||
| Allgemeines | |||||||||||||||||||
| Name | 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosylbromid | ||||||||||||||||||
| Andere Namen |
| ||||||||||||||||||
| Summenformel | C14H19BrO9 | ||||||||||||||||||
| Kurzbeschreibung |
weißes bis schwach gelbliches[1] Kristallpulver | ||||||||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||||||||
| |||||||||||||||||||
| Eigenschaften | |||||||||||||||||||
| Molare Masse | 411,20 g·mol−1 | ||||||||||||||||||
| Aggregatzustand |
fest | ||||||||||||||||||
| Schmelzpunkt | |||||||||||||||||||
| Löslichkeit |
| ||||||||||||||||||
| Sicherheitshinweise | |||||||||||||||||||
| |||||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||||||||||||||

Vorkommen und Darstellung
In ihrer grundlegenden Publikation aus dem Jahr 1901[6][9] beschrieben Wilhelm Koenigs und Eduard Knorr die Synthese des von ihnen erstmals als „Acetobromglucose“ benannten 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromids aus Glucose und Acetylbromid in einer Reinausbeute von 58 %.

Den Einsatz des relativ teuren und unangenehm zu handhabenden Acetylbromids vermeidet die zweistufige Synthese von Emil Fischer[10], bei der Glucose zunächst mit Acetanhydrid und Natriumacetat zur Pentaacetylglucose (74 % Ausbeute) und diese anschließend mit Bromwasserstoffsäure in quantitativer Ausbeute[11] bzw. mit einer gesättigten Lösung von Bromwasserstoff in Eisessig in 76%iger Ausbeute zur Acetobromglucose umgesetzt wird.

Bei der Reaktion von Glucose mit Acetanhydrid, das mit gasförmigem Bromwasserstoff gesättigt wurde, werden ebenfalls Reinausbeuten an Acetobromglucose von 50 bis 60 % erzielt.[12]
Die aus den frühen Synthesen von Acetobromglucose entwickelte Standardvorschrift[4] verläuft ebenfalls über Pentaacetylglucose, die mit gasförmigem Bromwasserstoff in einer Rohausbeute von 80 bis 87 % zum Endprodukt reagiert.
Die Verwendung von gasförmigem Bromwasserstoff vermeidet eine Reaktionsvariante, bei der Pentaacetylglucose in Chloroform zunächst mit rotem Phosphor und Brom gemischt wird unter intermediärer Bildung von Phosphortribromid, das anschließend durch Zugabe von Wasser zu Phosphonsäure und Bromwasserstoff reagiert. Die entstehende Acetobromglucose geht in die Chloroformphase über und wird in einer Ausbeute von 84 % (bezogen auf Pentaacetylglucose) isoliert.[13][14]

Eigenschaften
2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromid ist ein weißes bis schwach gelbes, geruchloses Kristallpulver und bildet beim Umkristallisieren aus Petrolether oder Diisopropylether „strahlig angeordnete, glänzende, weiße Nadeln“,[6] die sich in Wasser zersetzen und in vielen organischen Lösungsmitteln löslich sind. Acetobromglucose ist unter Lichtausschluss im Vakuumexsiccator über Natriumhydroxid monatelang stabil;[10] die Verbindung ist stark rechtsdrehend und reduziert Fehlingsche Lösung.[6] Bei einem Eluent der Zusammensetzung Dichlormethan:Etyhlacetat 2:1 hat es einen Rf-Wert von 0,70.[2]
Anwendungen
Substitutionsreaktionen an Acetobromglucose
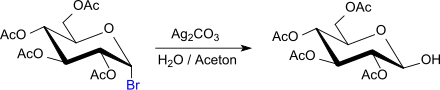
Die einfachste Reaktion des 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromids ist die Hydrolyse im Sinne eines nukleophilen Austauschs des Broms gegen eine Hydroxygruppe in Gegenwart von Silbercarbonat[15] zum β-D-Glucose-2,3,4,6-tetraacetat, das bei Reaktion in Aceton in Reinausbeuten von 75 bis 80 % anfällt.[16]
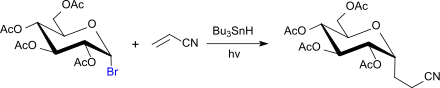
Im Gegensatz dazu handelt es sich bei der Reaktion der Acetobromglucose mit Acrylnitril in Gegenwart von Tributylzinnhydrid unter UV-Bestrahlung um eine radikalische Substitution, die zu 1-Deoxy-2,3,4,6-tetra-O-acetyl-1-(2-cyanoethyl)-α-glucopyranose führt.[17]
Der Austausch des Bromatoms in 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromid gegen Fluor mittels Kaliumhydrogendifluorid in Acetonitril liefert 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylfluorid in 70%iger Ausbeute,[18]

das sich wie Acetobromglucose als – allerdings weniger reaktiver – Glycosyldonor eignet.[19][20]
Aus 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromid ist über das Isothiuronium-Salz auf einfachem Wege 2,3,4,6-Tetra-O-acetyl-1-thio-β-D-glucopyranosid (peracetylierte β-Thioglucose) zugänglich, einem Schlüsseledukt für Auranofin, das zur Behandlung chronischer Polyarthritis eingesetzt wird.[21]

Nach Abspaltung der Acetylgruppen mit Natriummethanolat und Ansäuern wird 1-Thio-β-D-glucopyranosid erhalten.
Thioglycoside (statt H, R = Alkyl, Aryl) eignen sich ebenfalls als Glycosyldonoren (Aktivierung durch N-Iodsuccinimid/Silbertriflat) und zeichnen sich im Vergleich zu Glycosylhalogeniden wie Acetobromglucose durch deutlich höhere Stabilitäten aus.[22][23]
Acetobromglucose als Glycosyldonor
Bis zur Entdeckung der O-Glycosyl-trichloracetimidate[24][25] waren Glycosylhalogenide vom Typ des 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromids die Glycosyldonoren schlechthin.

Bereits im Jahr 1901 beschrieben W. Koenigs[6] und E. Fischer[11] die Darstellung des einfachsten β-Glucosids 2,3,4,6-Tetra-O-acetyl-β-methyl-D-glucopyranosid aus Acetobromglucose und Methanol mit unlöslichen Silbersalzen, z. B. Silbercarbonat, als Aktivatoren (Promotoren).

Als alternative Promotoren wurden von Burckhardt Helferich und Mitarbeitern Quecksilbersalze, wie z. B. Quecksilber(II)-cyanid Hg(CN)2 oder Quecksilber(II)-bromid HgBr2 bzw. Hg(CN)2/HgBr2-Gemische beschrieben,[26][27] die teilweise bessere Ausbeuten und Selektivitäten liefern, aber wegen der Giftigkeit der Quecksilber(II)-ionen und des bei der so genannten Helferich-Variante der Koenigs-Knorr-Methode entstehenden Cyanwasserstoffs weitgehend obsolet geworden sind.
Auch längerkettige Alkohole CnH2n+1OH mit n = 6–13 lassen sich in Ausbeuten um 60 % (nach Deacetylierung mit Natriummethanolat in Methanol) mit Acetobromglucose und Lithiumcarbonat als Promotor bei Raumtemperatur zu den entsprechenden n-Alkyl-β-D-glucopyranosiden umsetzen.[28]
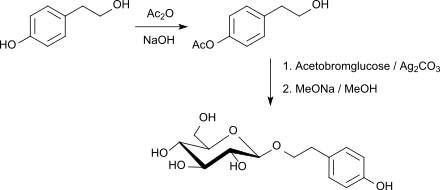
Pharmakologisch aktive Naturstoffe, wie z. B. Salidrosid mit antidepressiven und angstlösenden Eigenschaften sind durch Glycosylierung der alkoholischen Hydroxygruppe von Tyrosol (4-(2-Hydroxyethyl)phenol), einem Antioxidans aus Olivenöl, mit Acetobromglucose und Silbercarbonat als Promotor in einer Gesamtausbeute von 72 % nach Abspaltung der Acetylgruppen in Kilogrammmengen darstellbar.[29]
Acetobromglucose eignet sich auch zur Glucosylierung von Phenolen – in der wesentlich nukleophileren Phenolatform – z. B. von Hydrochinon zu Arbutin[30], von Salicylalkohol (Saligenin) zu Salicin oder von Vanillin zum 2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosylvanillin – in dieser Variante mit Tetrabutylammoniumbromid als besonders mildem Promotor in 50%iger Ausbeute.[31]

Eine weitere Abwandlung der Koenigs-Knorr-Methode zum Aufbau von Disacchariden aus 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosylbromid verwendet den aktivsten Promotor Silbertrifluormethansulfonat (Ag-triflat, AgOTf) in äquimolaren Mengen und als Protonenakzeptor Tetramethylharnstoff[32] Diese Verfahrensvariante zeichnet sich durch eine vereinfachte Prozessführung, sowie hohe Anomerenreinheit und Ausbeuten der Produkte aus.
Die Synthese komplexerer Oligosaccharide mit dem Glycosyldonor 2,3,4,6-Tetra-O-acetyl-α-D-glycopyranosylbromid wird durch seine thermische und chemische Instabilität und die Verwendung von teuren und toxischen Schwermetallsalzen als Promotoren, von Entwässerungsmitteln zur Bindung freiwerdenden Wassers bei Einsatz der unlöslichen Silbersalze Silber(I)-oxid und Silbercarbonat, sowie von Protonenakzeptoren zur Bindung des freiwerdenden Bromwasserstoffs eingeschränkt.[33][25]
Während die aktiven Hg- und Ag-Promotoren unter Nachbargruppenbeteiligung von Acylgruppen am Ringkohlenstoff C2 bevorzugt zu den kinetisch begünstigten β-Glycosiden (1,2-trans-Konformation) führen,
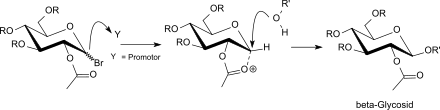
findet mit schwachen Promotoren, wie z. B. Tetraalkylammoniumhalogeniden (R4NBr) eine so genannte „in situ Anomerisierung“ unter Bildung der (reaktiveren) β-Acetobromglucose statt, die mit Glycosylakzeptoren zu α-verknüpften Glycosiden (1,2-cis-Konformation) führt.[34]
Acetobromglucose nimmt in der Aktivität als Glycosyldonor unter den peracetylierten Glycosylhalogeniden (Glycosyl-X) eine Mittelstellung ein: X: I > Br > Cl > F. Die Reaktivität von Glycosylakzeptoren sinkt von HO-Me >> HO-CH2-R > 6-OH >> 3-OH > 2-OH > 4-OH.[33]
Einzelnachweise
- Eintrag zu 2,3,4,6-Tetra-O-acetyl-D-glycopyranosyl Bromide (stabilized with CaCO3) bei TCI Europe, abgerufen am 25. August 2016.
- F. Zhang, A. Vasella: Regioselective synthesis of 1I, 1II, 5I, 5II, 6I, 6I, 6II, 6II-2H8-cellobiose. In: Carbohydrate Research. Band 342, Nr. 17, 2007, S. 2546–2556, doi:10.1016/j.carres.2007.07.017.
- Datenblatt Acetobromo-alpha-D-glucose bei Alfa Aesar, abgerufen am 25. August 2016 (Seite nicht mehr abrufbar).
- C.E. Redemann, C. Niemann: Acetobromoglucose [2,3,4,6-Tetraacetyl-α-d-glucopyranosyl bromide] In: Organic Syntheses. 22, 1942, S. 1, doi:10.15227/orgsyn.022.0001; Coll. Vol. 3, 1955, S. 11 (PDF).
- Pfaltz & Bauer: Acetobromo-a-D-glucose (Memento vom 1. Mai 2018 im Internet Archive), abgerufen am 25. August 2016.
- W. Koenigs, E. Knorr: Ueber einige Derivate des Traubenzuckers und der Galactose. In: Chem. Ber. Band 34, Nr. 1, 1901, S. 957–981, doi:10.1002/cber.190103401162.
- O. Lockhoff: Glycosylhalogenide. In: Hoeben-Weyl, Methoden der Organischen Chemie. 4. Auflage. E14a/3. G. Thieme, Stuttgart 1992, ISBN 978-3-13-776704-6, S. 626–728.
- V. Semeniuchenko, Y. Garazd, M. Garazd, T. Shokol, U. Groth, V. Khilya: Highly efficient glucosylation of flavonoids. In: Monatsh. Chem. Band 140, Nr. 12, 2009, S. 1503–1512, doi:10.1007/s00706-009-0207-6.
- A. Colley: Einige Bemerkungen zu der Abhandlung der HHrn. Koenigs und Knorr über Derivate des Traubenzuckers. In: Chem. Ber. Band 34, Nr. 2, 1901, S. 3205–3207, doi:10.1002/cber.190103402293.
- E. Fischer: Darstellung der Aceto-bromglucose. In: Chem. Ber. Band 49, Nr. 1, 1916, S. 584–585, doi:10.1002/cber.19160490162.
- E. Fischer, E. Frankland Armstrong: Ueber die isomeren Acetohalogen-Derivate des Traubenzuckers und die Synthese der Glucoside. In: Chem. Ber. Band 34, Nr. 2, 1901, S. 2885–2900, doi:10.1002/cber.190103402251.
- J.K. Dale: Preparation of bromoacetylglucose and certain other bromoacetyl sugars. In: J. Am. Chem. Soc. Band 38, Nr. 10, 1916, S. 2187–2188, doi:10.1021/ja.02267a030.
- R.U. Lemieux: Methods in Carbohydrate Chemistry, Vol. 2, Reactions of Carbohydrates. Hrsg.: R.L. Whistler, M.L. Wolfrom, J.N. BeMiller. Academic Press Inc., New York 1963, S. 221–222.
- K. Mohri et al.: Synthesis of glycosylcurcuminoids. In: Chem. Pharm. Bull. Band 51, Nr. 11, 2003, S. 1268–1272, doi:10.1248/cbp.51.1268.
- E. Fischer, K. Delbrück: Synthese neuer Disaccharide vom Typ der Trehalose. In: Chem. Ber. Band 42, Nr. 2, 1909, S. 2776–2785, doi:10.1002/cber.190904202203.
- C.M. McCloskey, G.H. Coleman: β-d-Glucose-2,3,4,6-tetraacetate (D-Glucose, β-2,3,4,6-tetraacetyl-) In: Organic Syntheses. 25, 1945, S. 53, doi:10.15227/orgsyn.025.0053; Coll. Vol. 3, 1955, S. 434 (PDF).
- B. Giese, J. Dupuis, M. Nix: 1-Deoxy-2,3,4,6-tetra-O-acetyl-1-(2-cyanoethyl)-α-D-glucopyranose In: Organic Syntheses. 65, 1987, S. 236, doi:10.15227/orgsyn.065.0236 (PDF).
- Patent US4751291: Process for the preparation of glycosyl fluorides protected on the oxygen. Angemeldet am 12. September 1986, veröffentlicht am 14. Juni 1988, Anmelder: Hoechst AG, Erfinder: J. Thiem, H.-M. Deger, W. Fritsche-Lang, M. Kreuzer.
- M. Yokoyama: Methods of synthesis of glycosyl fluorides. In: Carbohyd. Res. Band 327, Nr. 1–2, 2000, S. 5–14, doi:10.1016/S0008-6215(99)00324-9.
- K. Toshima: Glycosyl fluorides in glycosylations. In: Carbohyd. Res. Band 327, Nr. 1–2, 2000, S. 15–26, doi:10.1016/S0008-6215(99)00325-0.
- S. Pearson, W. Scarano, M.H. Stenzel: Micelles based on gold-glycopolymer complexes as new chemotherapy drug delivery agents. In: Chem. Commun. Band 48, 2012, S. 4695–4697, doi:10.1039/C2CC30510K.
- P. Konradsson, U.E. Udodong, B. Fraser-Reid: Iodonium promoted reactions of disarmed thioglycosides. In: Tetrahedron Lett. Band 31, Nr. 30, 1990, S. 4313–4316, doi:10.1016/S0040-4039(99)97609-3.
- G. Lian, X. Zhang, B. Yu: Thioglycosides in Carbohydrate Research. In: Carbohyd. Res. Band 403, 2015, S. 13–22, doi:10.1016/j.carres.2014.06.009.
- R.R. Schmidt, J. Michel: Einfache Synthese von α-und β-O-Glykosylimidaten; Herstellung von Glykosiden und Disacchariden. In: Angew. Chem. Band 92, Nr. 9, 1980, S. 763–765, doi:10.1002/ange.19800920933.
- R.R. Schmidt: Neue Methoden zur Glycosid- und Oligosaccharidsynthese – gibt es Alternativen zur Koenigs-Knorr-Methode? In: Angew. Chem. Band 98, Nr. 3, 1986, S. 213–236, doi:10.1002/ange.19860980305.
- B. Helferich, K.-F. Wedemeyer: Zur Darstellung von Glucosiden aus Acetobromglucose. In: Liebigs Ann. Chem. Band 563, Nr. 1, 1949, S. 139–145, doi:10.1002/jlac.19495630115.
- Patent DE19709787A1: Oligosaccharide und deren Derivate sowie ein chemo-enzymatisches Verfahren zu deren Herstellung. Angemeldet am 11. März 1997, veröffentlicht am 17. September 1998, Anmelder: Bayer AG, Erfinder: W.-D. Fessner, M. Petersen, A. Papadopoulos, G. Oßwald.
- V.Y. Joshi, M.R. Sawant: A convenient stereoselective synthesis of β-D-glucopyranosides. In: Ind. J. Chem. 45B, 2006, S. 461–465 (res.in [PDF]).
- T. Shi et al.: Development of a kilogram-scale synthesis of salidroside and its analogs. In: Synth. Commun. Band 41, Nr. 17, 2011, S. 2594–2600, doi:10.1080/00397911.2010.515332.
- C. Mannich: Ueber Arbutin und seine Synthese. In: Arch. Pharm. Band 250, Nr. 1, 1912, S. 547–560, doi:10.1002/ardp.19122500146.
- K. Mohri et al.: Synthesis of glycosylcurcuminoids. In: Chem. Pharm. Bull. Band 51, Nr. 11, 2003, S. 1268–1272, doi:10.1248/cbp.51.1268.
- S. Hanessian, J. Banoub: Chemistry of the glycosidic linkage. An efficient synthesis of 1,2-trans-disaccharides. In: Carbohydrate Research. Band 53, 1977, S. C13–C16, doi:10.1016/S0008-6215(00)85468-3.
- H. Paulsen: Fortschritte bei der selektiven chemischen Synthese komplexer Oligosaccharide. In: Angew. Chem. Band 94, Nr. 3, 1982, S. 184–201, doi:10.1002/ange.19820940304.
- R.U. Lemieux, K.B. Hendriks, R.V. Stick, K. James: Halide ion catalyzed glycosidation reactions. Syntheses of .alpha.-linked disaccharides. In: J. Am. Chem. Soc. Band 97, Nr. 14, 1975, S. 4056–4062, doi:10.1021/ja00847a032.