| Enfermedá de Creutzfeldt-Jakob | ||
|---|---|---|
 Modelu molecular del prion humanu PrPC. | ||
| Clasificación y recursos esternos | ||
| CIE-10 | A81.0, F02.1 | |
| CIE-9 | 046.1 | |
| OMIM | 123400 | |
| DiseasesDB | 3166 | |
| MedlinePlus | 000788 | |
| PubMed | Buscar en Medline mediante PubMed (en inglés) | |
| eMedicine | neuro/725 | |
| MeSH | D007562 | |
| Especialidá | neuroloxía | |
| [editar datos en Wikidata] | ||
La enfermedá de Creutzfeldt-Jakob (ECJ) ye una enfermedá neurolóxica con formes xenétiques hereditaries, producíes por una proteína llamada prion (PrP). Magar los casos hereditarios ya infeiciosos tán perfectamente documentaos, la causa de l'apaición del prion ye desconocida na mayor parte de los casos informaos. Trátase d'una enfermedá de naturaleza dexenerativa y pronósticu mortal qu'afecta aproximao a una persona por millón (prevalencia de 1:10⁶) a nivel global. N'Estaos Xuníos hai 127 pacientes de ECJ y les sos variantes (2006).
Acordies cola evidencia disponible, la ECJ resulta del plegamientu anormal d'un prion. Esti fenómenu paez aguiyar a qu'otres proteínes alterien les sos formes, afectando la so capacidá pa funcionar. Por esto, clasificar ente les enfermedaes priónicas o encefalopatías esponxiformes transmisibles (EET), caracterizaes por presentar una forma anómala de la proteína priónica celular (PrPC). Estes enfermedaes pueden esistir en formes esporádiques (idiopáticas), hereditaries, y adquiríes. El términu esponxiforme alude al aspeutu esponxosu que presenta na autopsia'l celebru afeutáu.
Xeneralidaes

La ECJ apaez xeneralmente na edá madura y evoluciona con rapidez, afectando en proporciones comparables a homes y muyeres.[1] Típicamente, los síntomes empiecen aproximao a la edá de 60 años y un 90 % de los pacientes muerre al cabu d'un añu. Nes etapes iniciales de la enfermedá, los enfermos sufren fallos de memoria, cambeos de comportamientu, falta de coordinación y perturbaciones visuales. A midida que progresa, el deterioru mental faise más pronunciáu y pueden dase movimientos involuntarios, ceguera, debilidá de les estremidaes y coma, rematando ensin esceición cola muerte del paciente.
El responsable de la ECJ ye un prion, partícula infeiciosa constituyida por una sola molécula de proteína, que nun contién acedos nucleicos nin información xenética, bien malo de destruyir ya inmune a los mecanismos d'esterilización tradicionales.
Descripción clásica
Anque ye posible que la enfermedá conocer dende la más remota antigüedá, los sos síntomes inespecíficos tienen de ser confundíos con otros tipos de llocura mientres sieglos. Esta enfermedá foi descrita per primer vegada polos neurólogos alemanes Hans-Gerhard Creutzfeldt y Alfons Maria Jakob en 1920. Dalgunos de los afayos clínicos qu'ellos describieron nos sos primeros documentos sobre la ECJ nun se correspuenden colos criterios actuales sobre la mesma, polo que se considera altamente probable que dalgunos de los casos estudiaos (a lo menos dos) nes sos investigaciones iniciales fueren víctimes d'otra enfermedá.
Los primeres síntomes de la enfermedá de Creutzfeldt-Jakob inclúin típicamente llocura —cambeos de personalidá xunto con deterioru de la memoria, el xuiciu y el pensamientu— y problemes de coordinación muscular. Les persones cola enfermedá tamién pueden esperimentar velea, depresión o sensaciones inusitadas. La ECJ nun causa fiebre nin otros síntomes comunes. A midida que progresa la enfermedá, el deterioru mental del paciente apínase. De cutiu empieza a tener contraiciones musculares involuntaries llamaes mioclono y puede quedar ciegu, perder el control de los esfínteres o una amplia variedá d'otros graves síntomes neurolóxicos. Col tiempu los enfermos yá nun pueden movese nin falar y cayen en coma. La neumonía y otres infeiciones compliquen de cutiu el cursu de la enfermedá y pueden conducir a la muerte por sigo mesmes.
La nueva variante
Alredor de 1920 describiéronse dellos casos que probablemente fueren en realidá de ECJ adquirida, anque munchos investigadores cunten que tratar d'otra enfermedá priónica denominada kuru, que se tresmitía ente los nativos de la etnia fore de Nueva Guinea por cuenta de los ritos funerarios de canibalismu familiar.
En 1996, reportar nel Reinu Xuníu la "nueva variante" de ECJ (ECJv),[2] y describiéronse diez caso asocedíos ente 1994 y 1995.[3] La nueva variante puede tresmitise por andada ente distintes especies y, posiblemente, de persona a persona. Anguaño postúlase que, en dellos casos, la forma adquirida de la ECJ sería la patoloxía humana subsiguiente a la infeición col llamáu "mal de la vaca lloca", nome con que se conoz la encefalopatía esponxiforme bovina (EEB). Sicasí, cabo esclariar que nel últimu casu, tantu la lleche, como'l músculu, el texíu adiposo y los fluyíos (cuspia, sangre, orina, semen) del ganáu bovino escarecen de capacidá infectiva per vía oral.[4]
Dende 1995 y hasta la metá del añu 2008 rexistráronse 204 casos d'esti tipu,[5] asocedíos mayoritariamente en Gran Bretaña.
Diagnósticu
El diagnósticu correutu de la ECJ ye bien difícil, porque de cutiu los síntomes pueden confundise colos d'otros trestornos neurolóxicos progresivos tales como'l Alzheimer o la enfermedá de Huntington. Sicasí, la ECJ causa inconfundibles cambeos nel texíu cerebral, claramente visibles na autopsia. Tamién tiende a causar un deterioru más rápidu de les capacidaes del paciente que la enfermedá d'Alzheimer o la mayoría de los demás tipos de llocura.
Na actualidá nun hai una prueba diagnóstica certera pa la enfermedá de Creutzfeldt-Jakob. Cuando un médicu abarrunta la presencia de ECJ, la primer esmolición consiste en refugar otres formes tratables de llocura tales como la encefalitis (inflamación del celebru) o la meninxitis crónica, polo que se riquir la evaluación por un neurólogu calificáu. Probar estándar de diagnósticu inclúin una punción espinal pa refugar otres causes de llocura y un electroencefalograma (EEG) pa rexistrar el patrón llétricu del celebru, que puede ser particularmente pervalible yá que amuesa un tipu específicu d'anomalía na ECJ.[1] La tomografía computarizada de celebru puede ayudar a refugar la posibilidá de que los síntomes sían la resultancia d'otros problemes tales como un ataque al corazón o un tumor cerebral. Les esploraciones del celebru por aciu imáxenes de resonancia magnética nuclear (RMN) tamién pueden poner de relieve patrón carauterísticos de dexeneración cerebral qu'ayuden a diagnosticar la ECJ.
La única forma de confirmar un diagnósticu de la ECJ ye por aciu una biopsia o autopsia cerebral. Nuna biopsia cerebral, el neurociruxanu dixebra un pequeñu cachu de texíu del celebru del paciente con cuenta de que pueda esaminalo un neuropatólogo. Esti procedimientu puede ser peligrosu pal paciente y l'operación non siempres llogra'l texíu de la parte afeutada del celebru. Por cuenta de que un diagnósticu correutu de la ECJ nun ameyora'l pronósticu nin les posibilidaes de tratamientu, la biopsia cerebral nun s'aconseya nun siendo que se precise pa refugar un trestornu tratable. Nuna autopsia, esamínase tol celebru dempués de la muerte.
Tipos
Hai tres clases principales de la enfermedá de Creutzfeldt-Jakob (ECJ):
ECJ esporádica
Nestos casos, la enfermedá preséntase entá cuando la persona paez tar llibre de factores de riesgu acomuñaos, esto ye, la etioloxía ye desconocida. El so algame ye mundial, siendo causada dacuando por una mutación ensin sentíu del xen de la proteína priónica (PRNP). Otres vegaes, l'avieyamientu ye l'únicu factor de riesgu consistente.[4] Coles mesmes, identificáronse otres mutaciones que nun causen direutamente la enfermedá pero vuelven a los individuos más susceptibles de contraer la infeición col prion. Estes últimes mutaciones taríen implicaes parcialmente na incidencia esporádica de la enfermedá.
Este ye'l tipu más común de ECJ, manifestándose en, siquier, un 85 % de los casos. Sicasí, nun ye posible adscribir direutamente los casos de ECJ esporádica a los otros dos grupos. La revisión de los afayos clínicos en casos de ECJv reveló qu'estos diferíen sustancialmente de los tradicionalmente reparaos en casos esporádicos.[6]
ECJ hereditaria
Puede determinase na hestoria del paciente dalgún antecedente familiar de la enfermedá o pruebes positives de mutación xenética acomuñada col xen productor del prion causante de la ECJ. Nos Estaos Xuníos, ente'l 5 y el 10 % de los casos de ECJ son d'orixe xenéticu y hereditariu. En 1950, reportóse y realizóse el siguimientu del primer casu familiar con miembros de tres generación probablemente afeutaos.[7] Coles mesmes, ta documentada la tresmisión de varón a varón. En 1979, establecióse que cerca d'un 15 % de los casos de ECJ son de tipu familiar.[8] En 1981, otru estudiu sobre 73 families determinó un historial consistente con un patrón d'heriedu autosómica dominante.[9] El fenotipu clínicu ye asemeyáu al reparáu nel ECJ esporádicu, anque esta forma suel presentase a edaes más tempranes.[4]
ECJ adquirida
La enfermedá ye tresmitida por esposición direuta al prion, por aciu contautu con texíos cerebrales o del sistema nerviosu infestaos. Probóse l'andada por aciu ciertos procedimientos médicos, tando tamién espuestos los veterinarios que tuvieron contautu con vaques o ovinos enfermos, personal de la industria de la carne, etc. Sicasí, nun hai pruebes de que la ECJ pueda arimase por aciu un contautu casual colos enfermos. Desque la ECJ describir por primer vegada, menos de 1 % de los casos probáronse como adquiríos más allá de toa dulda.
Magar la ECJ puede tresmitise de persona a persona, el riesgu de qu'esto asoceda ye por demás baxu. La ECJ nun paez poder tresmitise al traviés del aire o al tocar a otra persona o por aciu la mayoría de les formes de contautu casual. Los cónyuges y otros miembros de la familia de pacientes con ECJ esporádica nun tán sometíos a un riesgu mayor de contraer la enfermedá que la población polo xeneral (sacante nos casos obviamente hereditarios respectu de los fíos y otros descendientes).
- Forma iatrogénica
- El contautu direutu o indireutu col texíu cerebral y el líquidu del migollu espinal de los pacientes infestaos tien d'evitase pa torgar la tresmisión de la enfermedá al traviés d'estos materiales. Nunos cuantos casos bien raros pero perfectamente demostraos, la ECJ arrobinóse a otres persones arriendes d'inxertos de duramadre (una de les meninxes, texíos que cubren el celebru), córnees tresplantaes, implantación d'electrodos inadecuadamente esterilizados nel celebru y inyeiciones d'hormona somatotropa contaminada llograda de glándules pituitaries humanes tomaes de cadabres.[10][11] Los médicos llamen a estos casos provocaos por procedimientos médicos— "casos iatrogénicos".
- Nueva variante
- La enfermedá de Creutzfeldt-Jakob variante o nueva variante (ECJv o ECJnv) foi descrita nel Reinu Xuníu y en Francia, y empieza principalmente con síntomes psiquiátricos. Afecta a pacientes más nuevos que los d'otros tipos de ECJ y tien una duración más llarga de lo ordinario dende l'empiezu de los síntomes hasta la muerte. Foi afayada en 1996 y ye el tipu más rellacionáu cola esposición al prion responsable del mal de la vaca lloca. Créese que la ECJv ye adquirida a partir del ganáu infestáu con EEB.[12]
Bioloxía molecular
Nun principiu, creyóse que nel orixe de la ECJ y otres EET esistía un "virus lentu" (Lentivirus) o otru organismu desconocíu. Sicasí, estos nunca pudieron ser aisllaos. Amás, l'axente que causa la ECJ tien delles carauterístiques que son rares en microorganismos tales como los virus y les bacteries. Ye inmune a tolos métodos comunes d'esterilización, nun contién nenguna información xenética en forma d'acedos nucleicos (ADN o ARN) y presenta xeneralmente un llargu periodu d'incubación primero qu'apaezan los síntomes. En dellos casos, esti ralu puede ser d'hasta 40 años. La teoría científica principal —demostrada na actualidá pal kuru, la EEB y los casos adquiríos de ECJ— afirma qu'estes EET nun son causaes por un microorganismu sinón por un tipu de proteína llamáu prion.
Los priones presentar en forma normal como una proteína inocua topada nes célules del cuerpu, que controla ciertos aspeutos de la vida celular. Sicasí, el prion puede tomar tamién una forma infeiciosa capaz de causar la enfermedá. Este ye'l motivu de que'l sistema inmune nun seya capaz de lluchar contra'l prion, yá que se trata d'una proteína propio, que la so guarda ye normal en toles célules del cuerpu.
Les formes inocuas ya infeicioses de la proteína y el prion son cuasi idéntiques, pero la forma infeiciosa adquier una configuración plegada distintu a la de la proteína normal.
Na ECJ adquirida, el prion ingresa al organismu al traviés del contautu con priones infeiciosos. Na ECJ hereditaria, el xen responsable de producir la proteína normal sufrió una mutación tal que namái ye capaz de producir la proteína patolóxico. Acasu la causa de la inesplicable forma esporádica seya que los priones normales tresformar por razones entá desconocíes— na versión infeiciosa de la proteína.
La carauterística más letal d'estos priones patolóxicos ye qu'anque haya unu solu d'ellos, esta única molécula ye capaz de "reconfigurar" a los sos similares normales, produciendo una especie de imparable reacción en cadena que dexa al organismu ensin molécules "sanes". Una vegada qu'apaecen, les proteínes de los priones anormales xúnense y formen fibres o acumuladures llamaes "plaques amiloides", que pueden trate al microscopiu. Les fibres y les plaques pueden empezar a depositase años primero qu'empiecen a apaecer los síntomes de les ECJ. Inda nun ta claro'l papel que desempeñen estes estructures na enfermedá o cómo pudieren afectar a los síntomes.
Nes EET hereditaries, identificáronse delles (hasta 20) mutaciones distintes nel xen de los priones. La mutación específica que s'atopa en cada familia afecta posiblemente al tipu de EET que va esperimentar, a la frecuencia con qu'apaez la enfermedá na familia y al tipu de síntomes más notables. Sicasí, non toles persones con mutaciones nel xen de los priones adquieren la ECJ. Esto indica que les mutaciones pueden puramente aumentar la susceptibilidá a la ECJ y que seique esistan otros factores entá desconocíos que tamién desempeñen un papel na enfermedá, ensin refugar l'andada per diversu víes.
Xenética
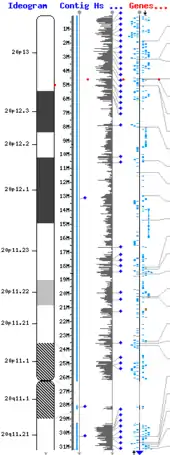
El xen productor del prion foi bautizáu PRNP (Prion-Related Protein). Esti xen atópase alcontráu nel brazu curtiu (p) del cromosoma 20, na posición 20pter-p12 (esto ye, ente la posición 20p12 y el final o términu del brazu), ocupando 15 000 pares de bases: más precisamente, dende'l Par de bases par 4 615 068 al 4 630 233 del cromosoma. La proteína codificada por esti xen denominar PrPC (c por "celular", yá que se topa presente en toles célules). Sicasí, una mutación nun únicu puntu d'esti xen puede producir una forma patolóxica de la mesma proteína, que foi llamada PrPSc (por scrapie, nome inglés de la tembladera ovina o caprina —ICTVdb 90.001.0.01.001).

El reemplazu d'una molécula del aminoácidu lisina por prolina n'unu de los sectores del xen PRNP fai qu'un grupu d'ocho aminoácido (octapéptidu) empiece a retrucar (copiase a sigo mesmu), diendo cada nueva copia a camudar a otres molécules de PrPC en PrPSc. D'esta miente esplícase la capacidá del prion pa convertir molécules normales en priones de la mesma.
En 1995 demostróse que l'error nesti únicu aminoácidu alteria radicalmente tola enorme estructura del restu de la proteína, reconfigurándola nuna nueva forma denominada prion.
Cinética molecular del prion
Al atopase una molécula de PrPsc con una normal, la "moldia" o repliega nuna forma distinta a la que tenía, reemplazando l'aminoácidu necesario pa convertila nuna como ella. Esta va tresformar a otres, y asina socesivamente. Esti procesu ye particularmente fácil y eficiente nes neurones. Parte de los túneles y vacuolas que dan al celebru enfermu'l so carauterísticu aspeutu d'esponxa son la resultancia del "estallíu" de neurones infestaes que lliberaron miles de priones nel mediu intercelular.
Les teoríes actuales afirmen que les célules nervioses tienen un receptor químicu na so membrana que se porta a manera de "pesllera". Ciertes proteínes "asesines", encargaes d'aniquilar a les neurones enfermes o anormales, introducen parte de la so estructura n'esti receptores a manera de "llave". El receptor de les célules sanes ta apexáu por una molécula de PrPc (esto ye, normal), de forma que les proteínes asesines nun puedan reconocela. Si'l furu ta llibre, la proteína asesino matu a la célula en cuestión.
Sicasí, el prion, cola so estructura deformada, nun puede ocupar el sitiu que-y correspuende na membrana, como una llave equivocada nun puede introducise nuna pesllera ayena. La célula normal, pos, presenta'l receptor llibre, dando a la proteína asesino la impresión de que se trata d'una que tien de morrer. Na ECJ y les demás EET, les proteínes asesines maten a les neurones normales confundiéndoles con célules marcaes pa ser destruyíes.
Enfermedaes rellacionaes
| EET o enfermedaes por priones n'animales[4] |
|---|
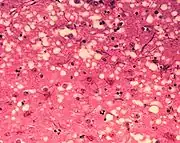 Microfotografía de texíu encefálico n'animal con EEB. y túneles que-y dan un aspeutu d'esponxa. |
| Enfermedá esporádica, posiblemente xenética ya infeiciosa |
| Scrapie (inglés), tremblante (francés), tembladera, mormeyera o rascadera (español) Cabres, oveyes, carneros |
| Enfermedá infeiciosa, posiblemente esporádica |
| EE bovina o "vaques lloques" (vaques y toros) |
| EE felina (gatos en llibertá y cautivos) |
| Encefalopatía transmisible del visón, del venáu y antílopes |
| EE de rumiantes de zoo y ungulaos |
| EE en diversos animales d'esperimentación |
| Enfermedá xenética |
| EE de mure trexénicu |
Esisten delles otres enfermedaes humanes rellacionaes cola ECJ, toes elles producíes por priones similares:
- Kuru: primer enfermedá priónica identificada nel ser humanu, de primeres confundir con una patoloxía virósica. Yera común nuna tribu aisllada de Nueva Guinea, ente que los sos habitantes acostumar comer los celebros de los familiares muertos. Foi afayada alredor de 1900, y, gracies a que los nativos fueron disuadidos de los sos estraños vezos, güei considerar estinguida.
- Síndrome de Gerstmann-Sträussler-Scheinker (GSS): tratar d'un mal perrraru, que solo afecta a un puñáu de families en tol mundu. Por esti motivu, considérase cuasi segura l'heriedu del xen responsable de producir el prion. De síntomes similares a la ECJ, el so desenvolvimientu ye más lentu (cursa ente 2 a 10 años). Ye tamién fatal ya incurable.
- Velea familiar fatal (IFF): producíu por una mutación distinta del xen del prion de la ECJ, ye desaxeradamente raru y los sos pocos casos rexistraos reparáronse n'España.
- Otres enfermedaes priónicas: la variedá de sintomatoloxíes clíniques nes enfermedaes priónicas llevó a la definición clínica d'otres patoloxíes del mesmu grupu (incluyendo numberosos casos d'Alzheimer que fueron reexaminados y güei cúntense ente les EET):
- Llocura per prion ensin patoloxía carauterística **
Llocura con paraparesia espástica
- Llocura talámica
- Encefalopatía esponxiforme familiar acomuñada a una nueva mutación nel xen PrP
- Gliosis subcortical progresiva **
Enfermedá mental ensin signos neurolóxicos
- Enfermedá d'Alzheimer familiar producida por priones
Coles mesmes, conócense numberoses variantes qu'afecten a distintes especies animales: équidos, bovinos, ovinos, caprinos, felinos, mustélidos y ungulaos monteses.
Tratamientu
Nun esiste dalgún tratamientu que pueda curar, ameyorar nin siquier controlar la sintomatoloxía na enfermedá de Creutzfeldt-Jakob. Los investigadores sometieron a prueba munchos fármacos, ente ellos l'amantadina, los esteroides, el interferón, el aciclovir, la clorpromazina,[13] y diversos axentes antivirales y antibióticos. Sicasí, nengunu d'estos tratamientos demostró ser beneficiosu.
L'únicu tratamientu posible de la ECJ tien como propósitu principal solliviar los síntomes hasta onde seya posible y ameyorar la calidá de vida del paciente. Les drogues opiáceas pueden ayudar a amenorgar el dolor si preséntase, y el clonazepam y el valproato de sodiu pueden ayudar a desaniciar el mioclono. Mientres les últimes etapes de la enfermedá, camudar de posición al paciente ayuda a evitar manques y escaras, mesmes de la postración en cama. Puede emplegase un catéter para drenar la orina si'l paciente nun puede controlar la función de la vexiga y tamién puede utilizase alimentación artificial, incluyendo líquidos intravenosos.
Investigación
Munchos investigadores tán estudiando la ECJ, tratando d'afayar los factores qu'inflúin na infectividad de los priones. Utilizando modelos de rucadores cola enfermedá y texíu cerebral d'autopsies, tamién tán intentando identificar aquellos factores qu'inflúin na susceptibilidá a la enfermedá y los que gobiernen el cursu de la mesma cuando apaez. Los estudios pa confirmar el diagnósticu son de bien alto costu y solo los países con teunoloxía de punta pueden realizalos, asina qu'entá ye más difícil faer el diagnósticu.
Esperen utilizar estes conocencies pa formular meyores pruebes diagnósticas y aprender el mecanismu íntimu pol que'l prion mata a les neurones con cuenta de que puedan formulase tratamientos eficaces.
El problema de les tresfusiones de sangre
Un informe científicu publicáu na prestixosa revista británica The Lancet[14] demostró que la ECJv puede tresmitise al traviés de les tresfusiones de sangre. El descubrimientu sollertó a los sistemes de salú, porque ye posible qu'una gran epidemia de la enfermedá apaeza nel futuru cercanu. Nun esiste una prueba que dexe determinar si'l sangre donada ta o non infestada col prion. Anque'l donante non presente síntomes, puede topase na fase latente de la enfermedá y tresmitila por aciu el sangre donada. Como reacción a esti informe, el gobiernu británicu prohibió donar sangre a toos aquellos que recibieren una tresfusión de sangre en fecha posterior a xineru de 1980.
El 28 de mayu de 2002 l'Alministración de Drogues y Alimentos de los Estaos Xuníos (FDA), prohibió de la mesma donar sangre a les persones que vivieren nes zones d'Europa consideraes d'altu riesgu de EEB o ECJ ente 1980 y mediaos de los 90. Dáu'l gran númberu de militares norteamericanos residentes n'Europa, créese que más del 7 % de los mesmos veránse impidíos de donar sangre en cumplimientu d'esta norma.
Midíes similares fueron adoptaes pola Cruz Roja Australiana, que nun dexa donar a quien vivieren un tiempu (acumuláu) de siquier seis meses nel Reinu Xuníu ente 1980 y 1996. Canadá prohibió la donación de sangre a les persones que vivieren seis meses o más nel Reinu Xuníu dende 1980. Lo mesmo aplícase a quien moraren en Francia por más de seis meses.
Víctimes famoses
La prensa masiva empezó a ocupase de la ECJ cuando finó víctima de la mesma'l célebre coreógrafu rusu-estauxunidense George Balanchine, a quien se considera la primer víctima famosa d'esti mal. L'artista nacíu en San Petersburgu empezó esperimentando estraños problemes d'equilibriu mientres danciaba en 1978. Dempués de perder la vista y l'oyíu, quedó dafechu incapacitado escontra 1982. Darréu desenvolvió anxina de pechu y un grave ataque cardiacu qu'obligó a practica-y una ciruxía de baipás coronariu. Ciegu, sordu y paralíticu, finó'l 30 d'abril de 1983. L'autopsia del so celebru demostró claramente que morriera de Creutzfeldt-Jakob.
Referencies
- López-Herrera A, et al. "El desafíu de les enfermedaes priónicas." Vet Méx. 2002;33(4). Disponible en llinia (PDF)
- McKintosh Y, Tabrizi SJ, Collinge J. "Prion diseases" Journal of NeuroVirology. 2003;9:183–93. Disponible en llinia (PDF)
- Notes
- 1 2 Brown P, Cathala F, Castaigne P, Gajdusek DC. "Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases." Ann Neurol. 1986 nov;20(5):597-602. PMID 3539001
- ↑ Cabo señalar que n'español dacuando utilízase esti términu de manera llibremente intercambiable cola espresión "Síndrome de Creutzfeldt-Jakob", lo que puede ser motivu de tracamundiu.
- ↑ Will RG, et al. "A new variant of Creutzfeldt-Jakob disease in the UK." Lancet. 1996 abr 6;347(9006):921-5. PMID 8598754
- 1 2 3 4 Bermejo FP, Muñoz D. "Encefalopatías esponxiformes transmisibles (EET) o enfermedaes producíes por priones." Rev Adm Sanitaria 2001;17:27-44.
- ↑ vCJD worldwide
- ↑ Tyler KL. "Creutzfeldt-Jakob disease." N Engl J Med. 2003 feb 20;348(8):711-9. PMID 12594311
- ↑ Jakob H, Pyrkosch W, Strube H. "Hereditary form of Creutzfeldt-Jakob disease (Backer family)." Arch. Psychiat. 1950;184(7):653-74. PMID 15433375
- ↑ Masters CL, Harris JO, Gajdusek DC, Gibbs CJ Jr, Bernoulli C, Asher DM. "Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familiar and sporadic clustering." Ann Neurol. 1979 feb;5(2):177-88. PMID 371520
- ↑ Masters CL, Gajdusek DC, Gibbs CJ Jr. "The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer's disease." Brain. 1981 sep;104(3):535-58. PMID 7023604
- ↑ Brown P, Preece MA, Will RG. "Friendly fire in medicine: hormones, homografts, and Creutzfeldt-Jakob disease." Lancet. 1992 jul 4;340(8810):24-7. PMID 1351607
- ↑ Brown P, Cervenakova L, Goldfarb LG, McCombie WR, Rubenstein R, Will RG, Pocchiari M, Martínez-Lage JF, Scalici C, Masullo C, et al. "Iatrogenic Creutzfeldt-Jakob disease: an example of the interplay between ancient genes and modern medicine." Neurology. 1994 feb;44(2):291-3. PMID 8309577
- ↑ Collinge J, Sidle KCL, Heads J, Ironside J, Hill AF. "Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD." Nature. 1996 oct 24;383(6602):685-90. PMID 8878476
- ↑ Korth C, et al. "Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease." Proc Natl Acad Sci U S A. 2001 ag 14;98(17):9836-41. PMID 11504948
- ↑ Peden AH, et al. "Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient." Lancet. 2004 ag 7-13;364(9433):527-9. PMID 15302196
Ver tamién
- Neuroloxía
- Encefalopatía esponxiforme bovina
- Prion
- Kuru
- Velea familiar fatal
- Encefalopatía esponxiforme familiar acomuñada a una nueva mutación nel xen PrP
- Alfons Maria Jakob
- Hans-Gerhard Creutzfeldt
Enllaces esternos
- Cronoloxía de la nueva variante de la enfermedá de Creutzfeldt-Jakob Por Joaquín Escudero Torrella. Seición de Neuroloxía. Hospital Xeneral de Castellón.
- Llocura vascular y otres enfermedaes que cursen con llocura Por F. Bermejo Pareja y otros. Medicine. 14 abr 2003;8(101):5453-64.
- Clínica de les enfermedaes priónicas Serviciu de Neuroloxía del Hospital Xeneral d'Elx.
- Sistema d'Información sobre Enfermedaes Rares
- Alzheimer Europe Información sobre l'Alzheimer, la ECJ y otros tipos de llocura.
- "Destructores de celebros", artículu informativu y didácticu sobre les enfermedaes producíes por priones
- N'inglés
- NCJDSU Unidá Nacional de Vixilancia de la ECJ del Reinu Xuníu.
- Genetics Home Reference Enfermedaes priónicas: variedá de recursos informativos; conteníos producíos polos National Institutes of Health (publicaos baxu dominiu públicu).
- Creutzfeldt-Jakob Disease Information Page NINDS
Plantía:MPN
| Control d'autoridaes |
|
|---|
 Datos: Q49989
Datos: Q49989 Multimedia: Creutzfeldt–Jakob disease
Multimedia: Creutzfeldt–Jakob disease