متلازمة ماكليود
متلازمة مكلاود هي مرض وراثي ينتقل بشكل متنح مرتبط بالكروموزوم الجنسي إكس، ويصيب الدم والدماغ والأعصاب المحيطية والعضلات والقلب. ينتج المرض عن عدد من الطفرات الموروثة بشكل مقهور، والمرتبطة بالجين إكس كي على الكروموزوم الجنسي إكس. يعد هذا الجين مسؤولًا عن إنتاج بروتين كي إكس، وهو بروتين ثانوي داعم للمستضد كيل على سطح الكريات الحمراء.
| متلازمة ماكليود | |
|---|---|
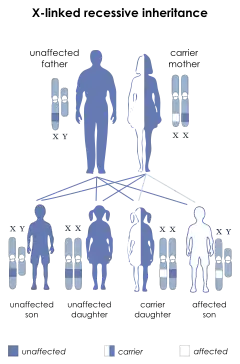 متلازمة مكلاود مرض وراثي ينتقل بصورة متنحية مرتبطة بالكروموزوم الجنسي إكس. متلازمة مكلاود مرض وراثي ينتقل بصورة متنحية مرتبطة بالكروموزوم الجنسي إكس. | |
| معلومات عامة | |
| من أنواع | داء الكريات الشائكة العصبي، ومرض الجهاز العصبي الوراثي التنكسي ، ومرض أيضي عصبي |
| الإدارة | |
| حالات مشابهة | متلازمة سوير–جيمس |
التظاهرات
يبدأ المرضى بملاحظة الأعراض عادة عند الوصول إلى عمر الخمسين، ويتطور المرض عادة على نحو بطيء. تشمل التظاهرات الشائعة للمرض الاعتلال العصبي المحيطي واعتلال العضلة القلبية وفقر الدم الانحلالي. من التظاهرات الأخرى غير الشائعة نذكر الحركات الرقصية للأطراف والعرات الوجهية والحركات الفموية الأخرى (مثل عض الشفتين واللسان) والنوبات الاختلاجية والخرف ذي البدء المتأخر والتبدلات السلوكية.
الوراثة
النمط الظاهري لوراثة متلازمة مكلاود هو طفرة متنحية على نظام زمرة الدم كيل. يعتبر جين مكلاود مسؤولًا عن تشفير بروتين إكس كي، وهو موجود على الكروموزوم الجنسي إكس،[1] ويملك الخصائص البنيوية لبروتين ناقل غشائي لكن وظيفته غير معروفة. يعتبر غياب البروتين إكس كي مرضًا وراثيًا مرتبطًا بالكروموزوم إكس.[2] تؤدي المتغيرات في هذه الطفرة إلى تنوع متلازمة مكلاود بين وجود داء الكريات الشائكة العصبي أو عدمه: الجين الموجود على الكروموزوم إكس والمسؤول عن متلازمة مكلاود قريب فيزيائيًا من الجين المسؤول عن داء الورم الحبيبي المزمن. نتيجة لذلك، يمكن أن يكون أي شخص مصاب بحذف جيني صغير نسبيًا مريضًا بهاتين المتلازمتين في الوقت ذاته.[3]
قد يكون النمط الظاهري موجودًا دون إصابة المريض بمظاهر هذه المتلازمة.[4]
التشخيص
الصفات المخبرية
تعتبر متلازمة مكلاود من الأمراض القليلة التي يمكن في سياقها ملاحظة الكريات الشائكة في لطاخة الدم المحيطي. يمكن أن يبدي التقييم الدموي علامات لفقر الدم الانحلالي. يمكن ملاحظة ارتفاع إنزيم كيناز الكرياتين في حالات الاعتلال العضلي في سياق متلازمة مكلاود.
الصفات الشعاعية والتشريحية المرضية
يظهر التصوير بالرنين المغناطيسي زيادة إشارة في الزمن الثاني (تي 2) لمنطقة البطامة الجانبية مع ضمور في النواة المذنبة وتوسع ثانوي للبطينين الجانبيين. يظهر تشريح جثث المصابين فقدانًا في الأعصاب وفسادًا دبقيًا عصبيًا في النواة المذنبة والكرة الشاحبة. قد تظهر تبدلات مشابهة في المهاد والمادة السوداء والبطامة. عادة ما تعف الإصابة عن المخيخ وقشر الدماغ.
العلاج
لا يوجد علاج شاف لمتلازمة مكلاود، ويعتمد تدبير المتلازمة على العلاج العرضي بناء على الأعراض. قد تفيد الأدوية في تدبير النوبات الاختلاجية والإصابات القلبية والنفسية، على الرغم من أن استجابة الحركات الرقصية على العلاج تكون سيئة عادة.[بحاجة لمصدر]
الإنذار
عادة ما يعيش المريض النموذجي المصاب بمتلازمة مكلاود التي تبدأ خلال البلوغ لمدة 5 إلى 10 سنوات. يحمل المرضى المصابون باعتلال العضلة القلبية خطورة أعلى للإصابة بقصور القلب الاحتقاني والموت القلبي المفاجئ. عادة ما يكون الإنذار جيدًا ومعدل الحياة طبيعيًا لدى المرضى المصابين بمضاعفات عصبية أو قلبية بسيطة.[5]
الوبائيات
تصيب متلازمة مكلاود بين 0.5 و1 من كل 100,000 شخص من مجموع السكان. يملك الذكور المصابون بالمتلازمة خلايا شائكة في الدم المحيطي بسبب آفة تصيب الوريقة الداخلية المزدوجة من غشاء كريات الدم الحمراء، بالإضافة إلى انحلال الدم الخفيف. تكون نسبة الكريات الشائكة أقل لدى الإناث وانحلال الدم خفيف جدًا؛ تفسر الإصابة الأخف لدى الإناث بتثبيط عمل الكروموزوم إكس عبر تأثير ليون. يطور بعض المصابين بنمط مكلاود الظاهري اعتلالًا عضليًا أو اعتلالًا عصبيًا أو أعراض نفسية، ما يؤدي إلى متلازمة قد تقلد الرقص.[6][7]
يمكن أن تسبب متلازمة مكلاود ارتفاعًا في إنزيمات كيناز الكرياتين (سي كي) ونازع هيدروجين اللاكتات (إل دي إتش)، الأمر الذي قد يلاحظ مصادفة عند إجراء التحاليل المخبرية الروتينية.[8]
التاريخ
اكتشفت متلازمة مكلاود عام 1961 وحمل هذا المرض اسم أول مريض تؤكد إصابته به بشكل مشابه لتسمية نظام مستضد كيل. لوحظ في فحص عينة دم مخصصة للتبرع قدمها طالب طب الأسنان في جامعة هارفارد هيو مكلاود تعبير ضعيف لمستضدات نظام كيل على سطح الكريات الحمراء، كما أبدى الفحص تحت المجهر ظهور كريات شائكة.[9]
تظهر زوجات الملك الإنجليزي هنري الثامن نمطًا مميزًا من الحمول ووفيات الأطفال حول الولادة، والذي يقترح عند ربطه بتراجع الحالة العقلية للملك أنه كان إيجابي المستضد كيل وعانى من متلازمة مكلاود.[10]
المراجع
- Arnaud L، Salachas F، Lucien N، وآخرون (مارس 2009). "Identification and characterization of a novel XK splice site mutation in a patient with McLeod syndrome". Transfusion. ج. 49 ع. 3: 479–84. DOI:10.1111/j.1537-2995.2008.02003.x. PMID:19040496.
- Ho MF، Monaco AP، Blonden LA، وآخرون (فبراير 1992). "Fine mapping of the McLeod locus (XK) to a 150-380-kb region in Xp21". Am. J. Hum. Genet. ج. 50 ع. 2: 317–30. PMC:1682457. PMID:1734714.
- Marsh WL، Oyen R، Nichols ME، Allen FH (فبراير 1975). "Chronic granulomatous disease and the Kell blood groups". Br. J. Haematol. ج. 29 ع. 2: 247–62. DOI:10.1111/j.1365-2141.1975.tb01819.x. PMID:1191546.
- Walker RH، Danek A، Uttner I، Offner R، Reid M، Lee S (فبراير 2007). "McLeod phenotype without the McLeod syndrome". Transfusion. ج. 47 ع. 2: 299–305. DOI:10.1111/j.1537-2995.2007.01106.x. PMID:17302777.
- Hewer، E؛ Danek، A؛ Schoser، B. G.؛ Miranda، M؛ Reichard، R؛ Castiglioni، C؛ Oechsner، M؛ Goebel، H. H.؛ Heppner، F. L.؛ Jung، H. H. (2007). "Mc Leod myopathy revisited: More neurogenic and less benign". Brain. ج. 130 ع. Pt 12: 3285–96. DOI:10.1093/brain/awm269. PMID:18055495.
- Danek A، Rubio JP، Rampoldi L، وآخرون (ديسمبر 2001). "McLeod neuroacanthocytosis: genotype and phenotype". Ann. Neurol. ج. 50 ع. 6: 755–64. DOI:10.1002/ana.10035. PMID:11761473.
- Malandrini A، Fabrizi GM، Truschi F، وآخرون (يونيو 1994). "Atypical McLeod syndrome manifested as X-linked chorea-acanthocytosis, neuromyopathy, and dilated cardiomyopathy: report of a family". J. Neurol. Sci. ج. 124 ع. 1: 89–94. DOI:10.1016/0022-510X(94)90016-7. PMID:7931427. S2CID:27859436. مؤرشف من الأصل في 2021-03-19.
- Oechsner M, G. Winkler G, A. Danek A, "McLeod neuroacanthocytosis: An underdiagnosed syndrome?" International communication forum in human molecular genetics September 6, 1995 نسخة محفوظة 2020-09-28 على موقع واي باك مشين.
- Allen FH، Krabbe SM، Corcoran PA (سبتمبر 1961). "A new phenotype (McLeod) in the Kell blood-group system". Vox Sang. ج. 6 ع. 5: 555–60. DOI:10.1111/j.1423-0410.1961.tb03203.x. PMID:13860532.
- "Discovery News: Henry VIII's eccentricities possibly explained". مؤرشف من الأصل في 2018-09-18.
 بوابة طب
بوابة طب