متلازمة لانجر-جيديون
متلازمة لانجر-جيديون هو اضطراب وراثي جسمي غير شائع ناتج عن حذف مادة كروموسومية. يدعى اثنين من الأطباء الذين اضطلعوا بالبحوث الرئيسية في حالة في الستينيات. وعادة ما يتم التشخيص عند الولادة أو في مرحلة الطفولة المبكرة.
متلازمة لانجر-جيديون | |
|---|---|
 A person showing the typical features of Langer-Giedion syndrome A person showing the typical features of Langer-Giedion syndrome | |
| تسميات أخرى | Deletion 8q24.1, monosomy 8q24.1, trichorhinophalangeal syndrome type II (TRPS2), Langer-Giedion chromosome region (LGCR)[1][2] |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | مرض وراثي سائد ، ومتلازمة |
| المظهر السريري | |
| الأعراض | قصر القامة[3] |
الأعراض
الاعراض تشمل: صعوبات التعلم، قصر القامة، ملامح الوجه فريدة من نوعها، الرأس الصغيرة وتشوهات الهيكل العظمي بما في ذلك النمو العظمي من أسطح العظام.[4] وعادة ما يكون لدى الأفراد الذين يعانون من متلازمة لانجر-جيديون شعر فروة الرأس، والأذنين التي قد تكون كبيرة أو بارزة، والحاجبين واسعين، عيون عميقه مجموعة الأنف المصلي، الشفة العليا الضيقة الطويلة، والأسنان المفقودة.
 القدم اليمنى من شخص مع لانجر–Giedion متلازمة تظهر ملامح مميزة
القدم اليمنى من شخص مع لانجر–Giedion متلازمة تظهر ملامح مميزة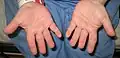 يد شخص مع لانجر–Giedion متلازمة عرض مميزة أصابع قصيرة.
يد شخص مع لانجر–Giedion متلازمة عرض مميزة أصابع قصيرة.
السبب
حذف 8q23.2 إلى q24.1.
وهو ينطوي على فقدان TRPS1 و EXT1.
التشخيص
يستند التشخيص على النتائج السريرية ويمكن تأكيده عن طريق اختبار الخلايا الوراثية، عندما يكون الحذف في المتوسط 5 ميغابايت (الملايين من أزواج القاعدة). في الوقت الحاضر هناك ممارسة شائعة لتشغيل (صفيف كروموسوم التهجين الجينوم) دراسة على الدم المحيطي للمريض، من أجل الحد من مدى فقدان المنطقة الجينومية، والجينات المحذوفة.[5]
العلاج
في حين لا يوجد متلازمة وراثية قادرة على الشفاء، والعلاجات المتاحة لبعض الأعراض. وقد استخدمت المثبتات الخارجية لإعادة بناء الحواف والوجه.
انظر أيضا
مراجع
- Disease Ontology (بالإنجليزية), 27 May 2016, QID:Q5282129
- Devidayal؛ Marwaha RK (فبراير 2006). "Langer-Giedion Syndrome" (PDF). Indian Pediatrics. ج. 43 ع. 2: 174–175. PMID:16528117. مؤرشف من الأصل (PDF) في 2017-05-16.
- OMIM Entry - # 150230 - TRICHORHINOPHALANGEAL SYNDROME, TYPE II; TRPS2 نسخة محفوظة 10 مارس 2017 على موقع واي باك مشين.
روابط خارجية
 بوابة طب
بوابة طب