متلازمة كيو تي الطويلة
متلازمة كيو تي الطويلة (بالإنجليزية: Long QT syndrome) هي اضطراب قلبي موروث نادر تتأخر بها عودة الاستقطاب في القلب مما يرفع احتمالات نوبات تسرع القلب البطيني مما قد يؤدي إلى ظهور نوبات لانظمية بطينية تدعى تورساد دي بوانت. هذه النوبات قد تؤدي إلى الخفقان، الإغماء والموت المفاجئ نتيجة لرجفان بطيني. هذه النوبات يمكن أن تكون مستثارة على يد عدة عوامل، حسب نميط المتلازمة.[2][3]
| متلازمة كيو تي الطويلة | |
|---|---|
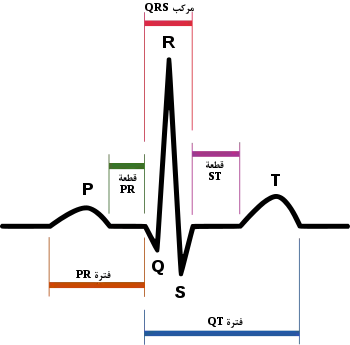 رسم توضيحي لنظم جيبي طبيعي لدى قلب إنساني كما يظهر في مخطط كهربية القلب (موسومة باللغة العربية) رسم توضيحي لنظم جيبي طبيعي لدى قلب إنساني كما يظهر في مخطط كهربية القلب (موسومة باللغة العربية) | |
| معلومات عامة | |
| الاختصاص | طب القلب |
| من أنواع | اضطراب نظم قلبي، واعتلال عضلة القلب الداخلي ، ومرض |
| المظهر السريري | |
| الأعراض | اضطراب نظم قلبي[1] |
| الإدارة | |
| أدوية | |
وراثيات
جينات وطفرات
متلازمة كيو تي الطويلة ممكن أن تنبع من طفرة في واحد من عدة جينات.[4] هذه الطفرات تؤدي إلى استطالة فترة جهد الفعل البطيني، مما يؤدي إلى استطالة فترة كيو تي. ممكن أن تورث المتلازمة بصيغة وراثة صبغية جسدية سائدة أو بصيغة وراثة صبغية جسدية متنحية.[5]
| نمط | OMIM | طفرة | ملاحظات |
| 1 | 192500 | وحيدة ألفا من قناة البوتاسيوم المقوّمة البطيئة الآجلة[6] (KvLQT1 أو KCNQ1) | |
| 2 | 152427 | وحيدة ألفا من قناة البوتاسيوم المقوّمة السريعة الآجلة (hERG أو MiRP1) | التيار عبر هذه القناة يعرف بـ Ikr. النمط الظاهري ناتج على الأغلب من تقلّص في تيار عودة الاستقطاب. |
| 3 | 603830 | وحيدة ألفا من قناة الصوديوم Nav1.5 (الجين هو SCN5A) | |
| 4 | 600919 | بروتين مثبّت Ankyrin B | |
| 5 | 176261 | وحيدة بيتا Mink (أو KCNE1) والتي تشترك في تكوين القناة مع KvLQT1 | |
| 6 | 603796 | وحيدة بيتا MiRP1 (أو KCNE2) والتي تشترك في تكوين القناة مع hERG | |
| 7 | 170390 | قناة البوتاسيوم KCNJ2 (أو Kir2.1) | متلازمة كيو تي الطويلة نمط 7 تؤدي إلى متلازمة أندرسن-طويل. |
| 8 | 601005 | وحيدة ألفا من قناة الكالسيوم Cav1.2 (الجين هو CACNA1c) | يؤدي إلى متلازمة تيموثي. |
| 9 | 611818 | Caveolin 3 | |
| 10 | 611819 | SCN4B | |
| 11 | 611820 | AKAP9 | |
| 12 | 601017 | SNTA1 | |
| 13 | 600734 | GIRK4 |
متلازمة كيو تي الطويلة المحدّثة بالدواء عادة ما يكون نابع من علاج بأدوية مضادة اضطراب النظم، مثل أميودارون أو سوتالول أو عدة أدوية أخرى تم التقرير عنها كمسببة لذلك (مثل سيسابريد). بعض مضادات الذهان، مثل هالوبيريدول وزيبراسيدون، تؤدي إلى متلازمة كيو تي الطويلة كواحد من أعراضها الجانبية النادرة. طفرات جينية ممكن تؤدي إلى وضع أن الشخص هو أكثر عرضة لمتلازمة كيو تي الطويلة المحدّثة بالدواء.
متلازمة كيو تي الطويلة نمط 1
النمط 1 من المتلازمة هو الأكثر انتشارًا[6]، حيث يشكل ما بين 30% و35% من حالات متلازمة كيو تي الطويلة. الجين المسبب للنمط الأول هو KCNQ1 المتواجد على الذراع القصيرة من صبغي 11 في الموضع الصبغي 15.5. الجين KCNQ1 يرمّز لقناة بوتاسيوم حسّاسة لفرق الجهد KvLQT1 ذات تعبير عالي في القلب. يظن العلماء أن ناتج الجين KCNQ1 ينتج وحيدة ألفا تتفاعل مع بروتينات أخرى (بالتحديد، وحيدة بيتا minK) لتكوّن القناة الأيونية IKs، المسؤولة عن تيار البوتاسيوم المقوّم البطيء الآجل في جهد الفعل القلبي.
متلازمة كيو تي الطويلة نمط 2
النمط 2 من المتلازمة هو الثاني الأكثر انتشارًا، حيث يشكل ما بين 25% و30% من جميع حالات متلازمة كيو تي الطويلة. الجين المسبب للنمط الثاني هو hERG المتواجد على الصبغي 7.[2]
متلازمة كيو تي الطويلة نمط 3
النمط 3 من المتلازمة نابع من طفرات في الجين المرمّز لوحيدة ألفا من قناة الصوديوم Nav1.5. هذا الجين هو SCN5A المتواجد على الذراع القصيرة من صبغي 3 في الموضع الصبغي 24-21.
وبائيات
متلازمة كيو تي الطويلة الموروثة تصيب حوالي 1 من كل 7,000 شخص.[7]
تشخيص
تشخيص متلازمة كيو تي الطويلة هو ليس عملية سهلة حيث أن 2.5% من الأشخاص المعافين لديهم كيو تي طويل، و10-15% من مرضى متلازمة كيو تي الطويلة لديهم فترة كيو تي سوية.[8] من هنا، معيار شائع جدًا لتشخيص متلازمة كيو تي الطويلة هي الحرز التشخيصي لمتلازمة كيو تي الطويلة.[9] يتم حساب الحرز التشخيصي بواسطة منح نقاط مختلفة لعدة معايير (مصنفة أدناه). عند الحصول على أربع نقاط أو أكثر، الاحتمال لوجود المتلازمة مرتفع; عند الحصول على نقطة واحدة أو أقل، الاحتمال لوجود المتلازمة مرتفع. حرز نقطتين أو ثلاث يدل على احتمال متوسط.[6]
- فترة كيو تي مصححة (معرّفة كحاصل قسمة فترة QT على جذر فترة RR)
- أكبر من 480 ميليثانية - 3 نقاط
- 470-460 ميليثانية - نقطتان
- 450 ميليثانية وجنس ذكر - نقطة واحدة
- لانظمية بطينية تدعى تورساد دي بوانت واضطرابات نظم دقات القلب البطيني - نقطتان
- موجة T متناوبة - نقطة واحدة
توقعات سير المرض
لدى مرضى متلازمة كيو تي الطويلة غير المعالجين، يمكن تنبؤ الاحتمال لحصول حوادث الإغماء أو الموت المفاجئ عن طريق النمط الجيني (متلازمة كيو تي الطويلة 1-8)، الجنس وطول فترة كيو تي المصححة.[10]
- اختطار مرتفع (أكبر من 50%)
فترة كيو تي مصححة أطول من 500 ميليثانية، متلازمة كيو تي الطويلة 1,2,3 (الذكور)
- اختطار متوسط (30-50%)
فترة كيو تي مصححة أطول من 500 ميليثانية، متلازمة كيو تي الطويلة نمط 3 (الإناث)
فترة كيو تي مصححة أقصر من 500 ميليثانية، متلازمة كيو تي الطويلة نمط 2 (الإناث)، متلازمة كيو تي الطويلة نمط 3
- اختطار منخفض (أصغر من 30%)
فترة كيو تي مصححة أقصر من 500 ميليثانية، متلازمة كيو تي الطويلة نمط 1 و2 (الذكور)
إمكانيات علاج
يُنصح الأشخاص المشخصون بمتلازمة كيو تي الطويلة بالامتناع عن تعاطي أدوية قد تطوّل فترة كيو تي أو ترفع احتمال حدوث اللانظمية البطينية تورساد دي بوانت.[11] بالإضافة إلى ذلك، هناك إمكانيتا علاج للمشخصين بمتلازمة كيو تي الطويلة: منع الاضطرابات النظمية وإيقاف الاضطرابات النظمية.
منع الاضطرابات النظمية
تُمنع الاضطرابات النظمية عن طريق استخدام الأدوية أو عمليات جراحية تهاجم سبب الاضظرابات النظمية المرتبطة بمتلازمة كيو تي الطويلة.
تاريخ
وصف الطبيب مايسر الحالة الموثقة الأولى لمتلازمة كيو تي الطويلة في لايبزيغ عام 1856، حيث توفيت فتاة صمّاء بعدما صرخ عليها معلمها. عندما وصفوا لأهلها تداعيات موتها، قال الأهل أن أخيها الأكبر والذي كان أصمّ أيضًا توفي بعد حالة فزع.[12]
انظر أيضًا
المراجع
- Disease Ontology (بالإنجليزية), 27 May 2016, QID:Q5282129
- Shinnawi R, Gepstein L (سبتمبر 2014). "iPS Cell Modeling of Inherited Cardiac Arrhythmias". Curr Treat Options Cardiovasc Med. ج. 16 ع. 9: 331. PMID:25080030. مؤرشف من الأصل في 2019-12-05.
- Morita H, Wu J, Zipes DP (أغسطس 2008). "The QT syndromes: long and short". Lancet. ج. 372 ع. 9640: 750–63. DOI:10.1016/S0140-6736(08)61307-0. PMID:18761222.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Hedley PL؛ Jorgensen P؛ Schlamowitz S؛ Wangari، Romilda؛ وآخرون (2009). "The genetic basis of long QT and short QT syndromes: a mutation update". Human Mutation. ج. 30 ع. 11: 1486–511. DOI:10.1002/humu.21106. PMID:19862833.
- Goldman 2011، صفحات 185
- Levine E, Rosero SZ, Budzikowski AS, Moss AJ, Zareba W, Daubert JP (أغسطس 2008). "Congenital long QT syndrome: considerations for primary care physicians". Cleve Clin J Med. ج. 75 ع. 8: 591–600. PMID:18756841. مؤرشف من الأصل في 2020-03-12.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Goldman 2011، صفحات 366
- Moric-Janiszewska E, Markiewicz-Łoskot G, Loskot M, Weglarz L, Hollek A, Szydlowski L (2007). "Challenges of diagnosis of long-QT syndrome in children". Pacing Clin Electrophysiol. ج. 30 ع. 9: 1168–1170. DOI:10.1111/j.1540-8159.2007.00832.x. PMID:17725765.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Schwartz PJ, Moss AJ, Vincent GM, Crampton RS (1993). "Diagnostic criteria for the long QT syndrome. An update". Circulation. ج. 88 ع. 2: 782–4. DOI:10.1161/01.CIR.88.2.782. PMID:8339437.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Ellinor PT, Milan DJ, MacRae CA (2003). "Risk stratification in the long-QT syndrome". N. Engl. J. Med. ج. 349 ع. 9: 908–9. DOI:10.1056/NEJM200308283490916. PMID:12944579.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - "QT Drug List by Risk Groups". Arizona Center for Education and Research on Therapeutics. مؤرشف من الأصل في 2013-03-27. اطلع عليه بتاريخ 2010-07-04.
- Tranebjaerg L؛ Bathen J؛ Tyson J؛ وآخرون (1999). "Jervell and Lange-Nielsen syndrome: a Norwegian perspective". American Journal of Medical Genetics. ج. 89 ع. 3: 137–46. DOI:10.1002/(SICI)1096-8628(19990924)89:3<137::AID-AJMG4>3.0.CO;2-C. PMID:10704188.
{{استشهاد بدورية محكمة}}: الوسيط غير المعروف|author-separator=تم تجاهله (مساعدة)
| داء قلبي إقفاري (نقص التروية) |
| ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| الطبقات |
| ||||||||||||||||||||||
| التوصيل/ اضطراب نظم قلبي |
| ||||||||||||||||||||||
| أخرى | تليف قلبي · ضخامة القلب · تضخم البطين (الأيسر، الأيمن) · ضخامة الأذين (الأيمن، الأيسر) قصور القلب (ربو قلبي) · حمى الروماتزم | ||||||||||||||||||||||
 بوابة طب
بوابة طب