متلازمة غاردنر
متلازمة غاردنر، المعروفة أيضا باسم داء سلائل القولون والمستقيم العائلي،[1] هو نوع وراثي صبغي جسدي سائد من داء السلائل التي تتميز بوجود زوائد متعددة في القولون مع أورام خارج القولون.[2] الأورام التي تحدث خارج القولون يمكن أن تشمل: الأورام العظمية للجمجمة، سرطان الغدة الدرقية، الأكياس البَشَرانِيَّة والأورام الليفية.[3] تحدث الاورام الرباطية في 15٪ من المصابين.
| متلازمة غاردنر | |
|---|---|
| معلومات عامة | |
| الاختصاص | طب الجهاز الهضمي، وعلم الأورام، وعلم الوراثة الطبية |
| من أنواع | داء السلائل الورمي الغدي العائلي |
| الإدارة | |
| حالات مشابهة | متلازمة الاغتراب الوالدي |
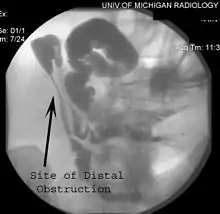
الأورام الرباطية هي الأورام الليفية التي تحدث عادة في الأنسجة التي تغطي الأمعاء ويمكن أن تثيرها عملية جراحية لإزالة القولون. سلائل القولون غير محدودة العدد عادة ما تسبق نشوء سرطان القولون. إذا لم تتم إزالة القولون، فإن فرصة الإصابة بسرطان القولون تكون كبيرة جدا. قد تنمو السلائل أيضا في المعدة والاثني عشر، الطحال، الكلى، الكبد، مساريق والأمعاء الدقيقة. في عدد قليل من الحالات، ظهرت السلائل أيضا في المخيخ. السرطانات المتعلقة بـهذه المتلازمة تظهر عادة في الغدة الدرقية والكبد والكلى. يزداد عدد السلائل مع التقدم في السن، ومئات الآلاف من السلائل يمكن أن تتطور في القولون.
تم وصف هذه المتلازمة لأول مرة في عام 1951.[4] وفي هذا الوقت، لا يوجد لها علاج. وفي أشكالها الأكثر تطورا، يُعتبر تشخيصها تشخيصا نهائيا مع توقع البقاء على قيد الحياة حتى عُمر 35-45 سنة والذي يعد متوسط العُمر المَأمُول؛ ويكون العلاج هو الجراحة والرعاية المُلطِّفة، وقد تم تجريب العلاج الكيميائي ولكن له نجاح محدود.[بحاجة لمصدر]
وراثيا
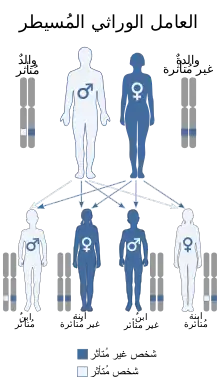
تورث متلازمة غاردنر كنمط الوراثة الصبغية الجسدية السائدة.[2] عادة، أحد الوالدين يكون لديه متلازمة غاردنر. كل واحد من أبنائهم، من الذكور والإناث على حد سواء، هو في خطر 50٪ من وراثة الجينات لمتلازمة غاردنر. ويزداد الخطر في كل جيل لاحق.
الأسباب
من المعروف ان متلازمة غاردنر سببها طفرة في جين داءُ السَّلاَئِلِ القُولُونِيُّ الوَرَمِيُّ الغُدِّيّ (APC gene) الموجود في كروموسوم 5q21, (الفرقة Q21 على كروموسوم 5).[2] وهذا هو نفس الجين الطّافِر (المتحوّر) في داء السلائل (داء البوليبات العائلي (FAP))، وهو مرض أكثر شيوعا ويهيئ للإصابة بسرطان القولون. وقد تسبب المعلومات الوراثية والجزيئية الجديدة بعض الاضطرابات الوراثية مما أدى إلى تقسيمها إلى كيانات متعددة إلا أن بعض الاضطرابات الوراثية الأخرى يتم جمعها في حالة واحدة. بعد وجودها لأكثر من النصف الثاني من القرن العشرين، اختفت متلازمة غاردنر ككيان منفصل. وتم دمجها في داء السلائل (داء البوليبات العائلي (FAP))، وتعتبر الآن اختلاف ظاهري من داء السلائل (داء البوليبات العائلي (FAP)).
التشخيص
تتكون متلازمة غاردنر من الزوائد (السلائل) الورميّة الغُديَّة في الجهاز الهضمي والأورام الرباطية، الأورام العظمية، الأكياس البَشَرانِيَّة، الأورام الشحمية، تشوهات الأسنان والسرطانات المُحِيْطٌة بالأَمْبُولَة. معدل حدوث هذه المتلازمة هو 1: 14025 لكلا الجنسين بالتساوي. يتم تحديد ذلك من قبل الجين الصبغي الجسدي السائد (APC) على الكروموسوم 5.[4]
يمكن التعرف على متلازمة غاردنر بناء على النتائج الفموية، بما في ذلك أثر انحشار الأسنان وزيادة عدد الأسنان (وهو جود أسنان زائدة أو إضافية للأسنان العادية)، الأورام العظمية المتعددة في الفك والتي تعطي مظهر «القطن والصوف» لمظهر الفكين، وكذلك الأورام السّنيّة المتعددة، وتضخم خلقي لظِهارَةٌ صِباغِيَّةٌ الشَبَكِيَّة (CHRPE)، بالإضافة إلى السلائل الورمية الغدية المتعددة في القولون. وترتبط متلازمة غاردنر أيضا مع داء السلائل (داء البوليبات العائلي FAP) وقد يظهر كـ وُرامٌ لِيفِيٌّ عُدْوانِيّ (الأورام الرباطية) في الحَيِّزُ خَلْفَ الصِّفاق.[5]
الأورام الرباطية تنشأ في معظم الأحيان من سِفاق العضلة المستقيمة البطنية في النساء متكررة الولادات. النوع الذي يحدث خارج البطن يكون نادر الحدوث ويمكن أن تنشأ أورام الثدي الرباطية في الغدد الثديية أو قد تحدث امتدادا لآفة ناشئة من عضلات جدار الصدر. الإصابة بأورام الثدي الرباطية تشكل ما هو أقل من 0.2٪ من أورام الثدي الأولية. في متلازمة غاردنر تتراوح الإصابة من 4٪ إلى 17٪. في الأورام الرباطية المرتبطة بمتلازمة غاردنر يكون هناك تغيير في مسار البيتا كاتينين (β-catenin) أو زيادة في افراز البيتا كاتينين (β-catenin ).[4]
مَسْماة (مصطلح يُنْسَب إلى اسم علم)
يُنسب اسم هذه المتلازمة إلى «الدون غاردنر» (1909-1989)، وهو عالم الوراثة الذي وصفها لأول مرة في عام 1951.[6]
المراجع
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN:1-4160-2999-0.
{{استشهاد بكتاب}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - الوراثة المندلية البشرية عبر الإنترنت (OMIM): 175100
- Luba MC, Bangs SA, Mohler AM, Stulberg DL (فبراير 2003). "Common benign skin tumors". Am Fam Physician. ج. 67 ع. 4: 729–38. PMID:12613727. مؤرشف من الأصل في 2008-05-16.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Rammohan A, Wood JJ (2012). "Desmoid tumour of the breast as a manifestation of Gardner's syndrome". Int J Surg Case Rep. ج. 3 ع. 5: 139–142. DOI:10.1016/j.ijscr.2012.01.00. PMC:3312056. PMID:22370045. مؤرشف من الأصل في 2015-09-24.
- DeVita: Cancer, Principles and Practice of Oncology, 8th Ed.p1742
- Gardner EJ (يونيو 1951). "A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum". Am. J. Hum. Genet. ج. 3 ع. 2: 167–76. PMC:1716321. PMID:14902760.