عوز البروتين سي
يعد عوز البروتين سي سمة وراثية نادرة تؤدي إلى الإصابة بالخثار. وُصف لأول مرة في عام 1981.[1] ينتمي المرض إلى مجموعة من الاضطرابات الوراثية المعروفة باسم أهبة التخثر (أو فرط الخثورية). يرتبط نقص البروتين سي بزيادة حدوث الجلطات الدموية الوريدية (الخطر النسبي 8-10)، بينما لم يُلاحظ وجود أي ارتباط بحدوث جلطات شريانية.[2]
| عوز البروتين سي | |
|---|---|
| Protein C deficiency | |
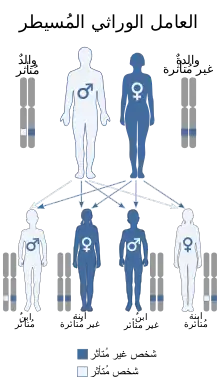 يُورث عوز البروتين سي بطريقة الوراثة الجسديَّة السائدة يُورث عوز البروتين سي بطريقة الوراثة الجسديَّة السائدة | |
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | مرض وراثي سائد ، وأهبة التخثر |
التظاهر
المضاعفات
يعتمد البروتين سي على فيتامين ك. المرضى الذين يعانون من نقص البروتين سي أكثر عرضة للإصابة بتنخر الجلد أثناء تناولهم الوارفارين. يتمتع البروتين سي بعمر نصفي قصير (8 ساعات) مقارنة بالعوامل الأخرى المعتمدة على فيتامين ك، وبالتالي يستنفذه الجسم بسرعة مع بدء استخدام الوارفارين، مما يؤدي إلى حالة فرط تخثر عابر.[3]
الفيزيولوجيا المرضية
تتمثل الوظيفة الرئيسية للبروتين سي في خصائصه المضادة للتخثر كمثبط لعوامل التخثر V و VIII. يؤدي عوزه إلى فقدان التحول الطبيعي للعوامل Va و VIIIa. يوجد نوعان رئيسيان من طفرات البروتين سي التي تؤدي إلى مرض عوز البروتين سي.[2]
- النمط الأول: عيوب كمية في البروتين سي (إنتاج منخفض أو عمر نصف قصير للبروتين)
- النمط الثاني: عيوب نوعية يكون فيها التفاعل مع الجزيئات الأخرى غير طبيعي. وُصفت عيوب في التفاعل مع الثرومبومودولين والفوسفوليبيد والعوامل الخامس والثامن وغيرها.
يفتقر غالبية الأشخاص الذين يعانون من نقص البروتين سي إلى نسخة واحدة فقط من الجينات الفعالة، وبالتالي فهم متغايري الزيجوت. قبل عام 1999، وُصفت ستة عشر حالة فقط من نقص البروتين سي متماثل اللواقح (نسختان غير طبيعيتين من الجين، مما أدى إلى عدم تصنيع بروتين سي فعال في مجرى الدم). قد يظهر هذا على شكل فرفرية فولمينان عند الأطفال حديثي الولادة.
التشخيص
يوجد نوعَين رئيسيين من مقايسات البروتين سي، التنشيط والمستضد (المقايسات المناعية). تعتمد فحوصات التنشيط المتاحة تجاريًا على الفحوصات المولدة للون التي تستخدم التنشيط بسم الثعبان في كاشف منشط، أو مقايسات الممتز المناعي المرتبط بالأنزيمات والتخثر. يسمح الاختبار المتكرر للنشاط الوظيفي للبروتين سي بالتمييز بين النقص العابر والخلقي للبروتين سي.[4][5]
في البداية، يمكن إجراء اختبار مقايسة نشاط البروتين C (الوظيفي)، وإذا كانت النتيجة منخفضة، فيمكن إجراء اختبار مقايسة مستضد البروتين سي لتحديد النوع الفرعي للنقص (النوع الأول أو النوع الثاني). في النمط الأول، تُصنَع جزيئات البروتين سي التي تعمل بشكل طبيعي بكميات منخفضة. في حالات النمط الثاني، يصنع الجسم كميات طبيعية من البروتين سي، لكنه يكون غير فعال.[6]
مقايسات المستضد هي مقايسات مناعية مصممة لقياس كمية البروتين سي بغض النظر عن وظيفتها. لذلك يتميز عوز النمط الأول بانخفاض في كل من النشاط وبروتين المستضد سي، بينما يتميز عوز النمط الثاني بمستويات طبيعية من مستضد البروتين سي، لكن مع مستويات نشاط منخفضة.[5]
يتكون جين البروتين سي البشري (PROC) من 9 إكسونات، وربط العلماء نقص البروتين سي بأكثر من 160 طفرة حتى الآن؛ لذلك لا يتوفر اختبار الحمض النووي لنقص البروتين سي بشكل عام خارج المعامل البحثية المتخصصة.[7]
يمكن ملاحظة ظهور فرفرية عابرة لأنها ترتبط عادة بتركيزات بروتين سي منخفضة في البلازما أقل من 5 ملغ وحدة دولية/ديسيلتر. التركيز الطبيعي لبروتين البلازما سي هو 70 نانومتر (4 ميكروغرام/مل) مع عمر نصف يبلغ حوالي 8 ساعات. ومع ذلك، فإن حديثي الولادة الأصحاء لديهم مستويات فيزيولوجية أقل (وأكثر تنوعًا) من البروتين سي (تتراوح بين 15-55 وحدة دولية/ديسيلتر) مقارنة بالأطفال الأكبر سنًا أو البالغين، وتزداد هذه التركيزات تدريجيًا طوال الأشهر الستة الأولى من الحياة. قد تكون مستويات البروتين سي أقل من 10 وحدة دولية/ديسيلتر عند الأطفال المولودين قبل أوانهم أو التوائم أو أولئك الذين يعانون من ضائقة تنفسية بدون إظهار فرفرية فولمينان أو تخثر منتشر داخل الأوعية.[8]
العلاج
غالبًا ما يوصف الهيبارين أو الوارفارين منخفض الوزن الجزيئي في الوقاية الأولية في الحالات العائلية المعروفة. يوصف العلاج الوقائي المضاد للتخثر لجميع الذين يصابون بجلطة وريدية بغض النظر عن السبب الكامن وراء ذلك. أظهرت الدراسات زيادة خطر حدوث الانصمام الخثاري الوريدي المتكرر في المرضى الذين يعانون من نقص البروتين سي. لذلك، يمكن وصف العلاج طويل الأمد بمضادات التخثر باستخدام الوارفارين لهؤلاء المرضى. يشكل عوز البروتين سي متماثل اللواقح مرضًا يحتمل أن يهدد الحياة، ويستدعي استخدام مركزات البروتين سي التكميلية. يمكن اعتبار زرع الكبد علاجيًا لنقص البروتين متماثل اللواقح.[9]
المراجع
- Griffin JH، Evatt B، Zimmerman TS، Kleiss AJ، Wideman C (1981). "Deficiency of protein C in congenital thrombotic disease". J. Clin. Invest. ج. 68 ع. 5: 1370–3. DOI:10.1172/JCI110385. PMC:370934. PMID:6895379.
- Khan S، Dickerman JD (2006). "Hereditary thrombophilia". Thromb J. ج. 4 ع. 1: 15. DOI:10.1186/1477-9560-4-15. PMC:1592479. PMID:16968541.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link) - "Protein C Deficiency". Cleveland Clinic. مؤرشف من الأصل في 2023-03-02. اطلع عليه بتاريخ 2023-02-24.
- Khor B، Van Cott EM (2010). "Laboratory tests for protein C deficiency". Am J Hematol. ج. 85 ع. 6: 440–442. DOI:10.1002/ajh.21679. PMID:20309856.
- Chalmers E، Cooper P، Forman K، Grimley C، Khair K، Minford A، Morgan M، Mumford AD (2011). "Purpura fulminans: recognition, diagnosis and management". Arch Dis Child. ج. 96 ع. 11: 1066–1071. DOI:10.1136/adc.2010.199919. PMID:21233082. S2CID:206846385.
- Rovida E، Merati G، D'Ursi P، Zanardelli S، Marino F، Fontana G، Castaman G، Faioni EM (2007). "Identification and computationally-based structural interpretation of naturally occurring variants of human protein C". Hum Mutat. ج. 28 ع. 4: 345–55. DOI:10.1002/humu.20445. PMID:17152060. S2CID:33496144.
- Williams MD، Chalmers EA، Gibson BE (202). "The investigation and management of neonatal haemostasis and thrombosis". Br J Haematol. ج. 119 ع. 2: 295–309. DOI:10.1046/j.1365-2141.2002.03674.x. PMID:12406062. S2CID:2022159.
- Manco-Johnson MJ، Abshire TC، Jacobson LJ، Marlar RA (1991). "Severe neonatal protein C deficiency: prevalence and thrombotic risk". J Pediatr. ج. 119 ع. 5: 793–798. DOI:10.1016/s0022-3476(05)80305-1. PMID:1834822. مؤرشف من الأصل في 2021-05-07.
- Kroiss S، Albisetti M (2010). "Use of human protein C concentrates in the treatment of patients with severe congenital protein C deficiency". Biologics: Targets and Therapy. ج. 4 ع. 5: 51–60. DOI:10.2147/btt.s3014. PMC:2846144. PMID:20376174.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link)
 بوابة طب
بوابة طب