ضمور العضلات الشوكي
ضمور العضلات الشوكي(إس إم إيه) (بالإنجليزية Spinal Muscular Atrophy -S.M.A)مرض وراثي يصيب الأعصاب التي تظهر من الحبل الشوكي الموجود في العمود الفقري.[1] ويظهر على شكل ضمور عضلات الأطراف مع ارتخاء شديد في العضلات. مرض ضمور العضلات الشوكي (إس إم إيه) يجعل العضلات في أجسام المصابين أضعف.وهذا يعني أن لدى الأشخاص المصابون بمرض (إس إم إيه)مشاكل تنفّسية ومشاكل في البلع. هناك عدة تقسيمات لهذا المرض.[2]
| ضمور العضلات الشوكي | |
|---|---|
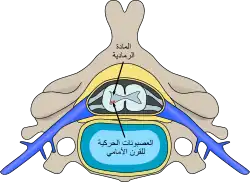 موقع الخلايا العصبية المتضررة من ضمور العضلات الشوكي في النخاع الشوكي موقع الخلايا العصبية المتضررة من ضمور العضلات الشوكي في النخاع الشوكي | |
| تسميات أخرى | autosomal recessive proximal spinal muscular atrophy |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | مرض العصبون المحرك، وضمور عضلي نخاعي، ومرض |
| الإدارة | |
| أدوية | |
الأنواع
| نوع | المسبب | عمر البداية | الخصائص | المراجع |
|---|---|---|---|---|
| شديد I-1 | داء ويردنج-هوفمان Werdnig-Hoffmann Disease | ستة أشهر-0 | قلة
حركة الجنين في بطن الأم خاصة في الأشهر الأخيرة من للحمل، ارتخاء وضعف في العضلات خاصة الأطراف والرقبة مع قلة في الحركة، وهذا الارتخاء والضعف يزداد مع مضي الوقت. كما قد تضعف عضلات البلع والمص فتظهر مشاكل في التغذية والبلع. وتكثر عمليات دخول الطعام إلى القصبة الهوائية عن الرضاعة. ويصعب على الطفل التحكم في بلع الريق والإفرازات المخاطية المتراكمة في الحلق. اهتزازات مستمرة في اللسان تجعل تشخيص المرض أكثر وضوحا لدى الأطباء. ويظهر لديه ضعف عام بعضلات ويبدو الصدر غائر وضيق من أعلى. كما يبذل مجهود كبير للتنفس بسبب ضعف وارتخاء عضلات القفص الصدري. ويمكن ملاحظة ذلك بتحرك عضلات البطن بدل الصدر عند التنفس وذلك ناتج لأن عضلات القفص الصدري ضامرة ولا تقوم بدورها ما عدى الحجاب الحاجز والذي يفصل الصدر عن البطن. ومع تدهور مشاكل التغذية والبلع والتنفس فإن الطفل تتدهور حالته إلى أن يتوفى في السنة الثانية من العمر. |
[3] |
| متوسط 2- II | ضمور العضلات الشوكي الوليدي المزمن | 6-18 شهر | ضعف في عضلات التنفس وأن كان يستطيع أن يتنفس بشكل جيد وكافي إلى أن قدرته على المحافظة على مستوى كافي من الأكسجين
خلال النوم صعب بعض الشيء وقد يحتاج الشخص إلى اسطوانة أكسجين كمساعدة. كما أن هناك ضعف في الكحة وطرد الإفرازات التنفسية ولذلك قد تكثر لديهم الالتهابات الرئوية. كما تظهر الاهتزازات المميزة لمرض ضمور العضلات الشوكي في اللسان في هذا النوع أيضا. انحناء وتقوس في العمود الفقري قد يستدعي إجراءعملية جراحية مع هشاشة عامة في العظام. نمو الجسم يزداد الضغط على الخلايا العصبية ولذلك قد تدهور صحة المريض مع الوقت.يستطيع الطفل المصاب بهذا النوع من الجلوس بدون وضع ساند له.مع أنه في الغالب يحتاج من يحرك له وضعية الجلوس عندما يكون مستلقي. في الغالب فان المصاب بهذا النوع لا يواجه مشاكل في التغذية، ولكن قد يصعب عليه تناول كمية كافية من الغذاء عن طريق الفم نتيجة لضعف المضغ والبلع. ولذلك قد يحتاج لتناول المزيد من الطعام والغذاء عبر أنبوب في الأنف أو عن طريق البطن مباشرة إلى المعدة، خاصة إذا كان هناك شك من دخول الطعام إلى الجهاز التنفسي. | [4] |
| خفيف 3-III | مرض كوجل بيرغ-فليندر (Kugelberg-Welander) | بعد 18 شهر | يستطيع المريض إن يقف ويمشي ويمارس حياته الطبيعية ومع عدم تأخر في النمو أو اكتساب المهارات الأساسية. ولكن قد يلاحظ الأهل كثر ة تعثر الطفل وصعوبة في النهوض من وضع الجلوس وفي لانحناء وقد يتطور المرض حتى يصبح المريض غير قادر على المشي مع تقدم العمر فيحتاج إلى استعمال الكرسي المتحرك.يتراوح سن ظهور الأعراض في هذا النوع بشكل كبير.فقد يتراوح ظهور الأعراض بين السنة الأولى من العمر إلى وقت البلوغ حتى بعد ذلك العمر.مع أن الأعراض في العادة تبدأ بالظهور خلال الثلاث السنوات الأولى.تبدأ الأعراض عادة في الأيدي، الأقدام واللّسان وانتشرت إلى المناطق الأخرى للجسم | ref>http://omim.org/entry/253400</ref> |
| الكبار IV-4 | سن البلوغ | تظهر أعراض هذا النوع من المرض بعد السن 35 سنة. ومن النادر جدا أن تظهر بين سن ثمان عشر سنة وثلاثون سنة.و تظهر الأعراض بالتدريج وبشكل بطيء كما أنها من النادر
أن تصيب عضلات الفم المتعلقة بالبلع وتنسيق التنفس. | [5] |
ومن الأشياء الأساسية والمميزة لمرض ضمور العضلات الشوكي بكل أنواعه، هو سلامة الحواس الأخرى وسلامة العقل والإدراك والتفكير. النوع العمر عند الإصابة العمر الافتراضي مهارات النمو إضافات النوع الأول قبل 6 أشهر اقل من سنتين يجلسون بالمساعدة صعوبات واضحة في التنفس والبلع. مع اهتزز في اللسان النوع الثاني 6-18 شهر 70% يعيشون إلى 25 سنة يستطيعون الجلوس اهتزاز في الأصابع واللسان.يصاب 50% بتقوس بالظهر، النوع الثالث بعد 12 شهر طبيعي يعتمدون على أنفسهم في الحركة النوع الرابع بالغ طبيعي طبيعية.[6][7][8][9]
الأسباب
مرض ضمور العضلات الشوكي ناتج عن خلل (طفرة), في جين يسمى اختصارا (إس إم أن- SMN).[10] والمعلومات المتوفرة في هذا الوقت هو أن هذا الجين ينتج بروتين له دور مهم في الخلايا الأمامية للحبل الشوكي.و التي تتحكم بحركة العضلات الموجودة في أجزاء الجسم المختلفة عن طريق ألياف عصبية طويلة. فعند رغبة الإنسان مثلا في تحريك عضلة معينة فإن الخلية الأمامية ترسل إشارة كهربائية عبر ألياف عصبية لتحرك العضلة. وعند ضمور الخلايا الأمامية فإن العضلات لا تستطيع الحركة ومع الوقت تضمر تلقائيا بسبب المرض. وبغض النظر عن الدور الحقيقي للبروتين المنتج من جين إس إم إن فان هذا البروتين مهم في استمرار الخلية الأمامية في أداء دورها.[11] ومع أن ضمور الخلايا الأمامية متواصل فالمصابين بالمرض فان العبء عليها أيضا يزداد مع نمو الجسم وزيادة العضلات وحاجة الجسم للحركة. ولذلك فان الأمر يزداد تعقيدا فمع عدم الحركة وضمور العضلات فإن الهشاشة في العظم تزداد ويبدأ العمود الفقري بالانحناء نتيجة لعدم أداء العضلات لدورها المتوقع والذي هو الحركة والحفاظ على توتر (شد) العضلات.[12][13]
الاعراض
تكون اعراض المرض بشكل عام في: o ضعف وارتخاء متزايد للعضلات (ضعف القدرة والقوة)
o ضمور متزايد للعضلات (نقص الحجم)
o قد تنتج عن ضمور العضلات تشوهات ثانوية في بعض المفاصل
o تأثر عضلات التنفس، وتأثر عضلات الوجه والبلع bulbar or brainstem muscles
o عدم وجود أعراض لإصابات الدماغ (النوبات التشنجية، فقدان التوازن، العجز الحسي)
o نقص ردّود أفعال الوترِ العميقة
o وجود التحزم fasciculations- وهي حركات ارتعاشية صغيرة للعضلات
o الذكاء - طبيعي
o تشبه أعراض الضمور العضلي أعراض الحثل العضلي مما قد يؤدي إلى صعوبة تشخيصه.
1-مرض وردنج هوفمان Werdnig-Hoffman disease - الضمور العضلي الشوكي الوليدي الحاد
o غالباً ما تظهر الأعراض من الولادة وحتى ستة أشهر
o 95%من المصابين تظهر عليهم الأعراض خلال ثلاثة أشهر
o ضعف شديد ومتزايد للعضلات مما يؤدي للارتخاء العام
o لا يستطيع الطفل التحكم في الرقبة
o ضعف عضلات الوجه والبلع والتنفس، مما يؤدي إلى ضعف القدرة على المص والبلع، وضعف القدرة على التنفس
o الضعف أشد في العضلات القريبة من العمود الفقري أكثر من العضلات الطرفية
o عدم تأثر عضلات العين (الحركة طبيعية)
o تشوهات ثانوية في بعض المفاصل، قد تظهر من الولادة
o الاختبارات الحسية سليمة وطبيعية
o نقص ردّود أفعال الوترِ العميقة
o وجود التحزم fasciculations- وهي حركات ارتعاشية صغيرة للعضلات، وقد تظهر على اللسان
o عدم وجود علامات لإصابة الدماغ
o أنتباه الطفل طبيعي
o نقص حركة الجنين في 30% في بعض الحالات
o الوفاة في عمر مبكر - 95% خلال 18 شهر
2-الضمور العضلي الشوكي الوليدي المزمن (SMA type II)وهي أكثر الأنواع شيوع:
o غالباً ما تظهر الأعراض بين 6-18 شهر
o تأخر التطور الحركي عن الوقت المتوقع لحدوثه: الجلوس، الوقوف، المشي.
o الضعف أشد في العضلات القريبة من العمود الفقري أكثر من العضلات الطرفية.
o وجود التحزم fasciculations-
o تضخم كاذب لعضلة بطة الساقgastrocnemius muscle.
o تشوهات جسمية
o صعوبات التنفس، الألتهابات التنفسية المتكررة (سبب رئيسي للوفاة ) o الاختبارات الحسية طبيعية
o نقص ردّود أفعال الوترِ العميقة
o المرض متزايد ومتطور، غالباً ما يعيش الطفل لعدة سنوات حسب الحالة.
3-مرض كوجيلبيرج ويلاندر Kugelberg-Welander disease- الضمور العضلي الشوكي للاحداث (الأطفال) (SMA III).
o ضعف العضلات يكون بشكل بطيء، ويؤثر أكثر على العضلات القريبة من الحبل الشوكي
o يبدأ الطفل في الحبو، الوقوف، المشي، ولكن يجد صعوبة في الحركات الكبيرة مثل الصعود والنزول من السلالم.
o تتأثر عضلات الأطراف السفلية أكثر من الأطراف العلوية مثل اليدين.
o ضعف عضلات الوجه والتنفس تحدث بشكل متأخر.
o عادة ما يكون مدى الحياة طبيعي
4-الضمور العضلي الشوكي للبالغين adult onset SMA - (SMA type IV)
o ضعف العضلات يكون بشكل بطيء.
o عادة ما يكون مدى الحياة طبيعي
التشخيص
في السابق كان الأطباء يعتمدون على عينة العضلات لتشخيص النهائي للمرض، ولكن مع توفر التحليل الجيني(تحليل SMN gene) قل بشكل كبير إجراء هذا التحليل ويجرى في المراكز التي لا يتوفر فيها هذا التحليل أو في الحالات التي ظهرت فيها نتائج التحليل سليمة بينما الأطباء لديهم قناعة في الإصابة بهذا المرض.[14]
يفحص الأطباء المرض حسب الأعراض الخارجية للمرض وتسلسل تاريخ المرض.[15] ومن أهم الأعراض هي ارتخاء وضعف العضلات واهتزاز اللسان. ويستعين الأطباء لاختبارات التشخيصية والتي تتمثل في:
o تحليل كرياتينين فوسفوكاينيزserum CPK طبيعي.
o التشخيص الوحيد يكون من خلال تحليل الكروموسومات.
o تخطيط الأعصاب الكهربائي Electrophysiologic ، يظهر ان الأعصاب الحسية سليمة، وأن المشكلة في الأعصاب الحركية.
o تخطيط العضلات الكهربائي (Electroyography)، ويظهر ان الضعف في حركة العضلة ناتج من الأعصاب وليس من العضلات [16]
o فحص عينة من العضلة تحت المجهرmuscle biopsy
o تخطيط القلب الكهربائي، ويكشف وجود أي نشاط غير طبيعي للقلب
حالات متشابهة التشخيص
يوجد العديد من الحالات، قد تحمل بعض العلامات المرضية المشابهة لمرض ضمور العضلات الشوكي، وتتبع أيضا بعض الاختبارات التشخيصية المتشابهة لها ولكنها تختلف عنها، ومنها: o الحثل العضلي - ضمور العضلات - Muscular dystrophy
o تصلب الانسجة الأولي الجانبي- Primary Lateral Sclerosis
o التصلب العصبي المتعدد -Multiple Sclerosis
o أمراض العضلات الخلقية- Congenital Myopathies
o الوهن العضلي- Myasthenia Gravis
o متلازمة داون- Down syndrome
o متلازمة هيرلر- Hurler syndrome
o متلازمة مارفان -Marfan syndrome
o متلازمة برادر ويلي- Prader-Willi syndrome
o مرض جوشير الولادي- Infantile Gaucher disease
o الأمراض الاستقلابية - bolic disorders
العلاج
حتى الآن ليس هناك علاج يمنع المرض من الحدوث أو يزيله، ولكن يتم العلاج لتقليل تأثيرات المرض على الطفل المصاب، ومنها: oالرعاية الغذائية: التغذية للرضيع عن طريق الأنبوبة التغذوية.[17][18][19]
oالرعاية التنفسية: منع الألتهابات الرؤية، وعلاجها.[20][21]
o الوقاية من المضاعفات في المفاصل والعظام.
o العلاج الطبيعي: والهدف منها تقليل التقفعات والعاهات وتاخير حدوثها، المحافظة على القوة العضلية، الحفاظ على أقصى جهد وظيفي، زيادة الحركة للمفاصل والوظيفة بواسطة الجبائر، الحفاظ على زيادة سعة التنفس.
o النشاط الرياضي: الخمول يساعد على زيادة الشد العضلي والتشوهات، لذلك ممارسة بعض الرياضة ولو كانت بالمساعدة من الاخرين تشجع العضلات على استعادة بعض لنشاط والحيوية بها.
o جراحة العظام: قد يفقد الطفل القدرة على المشي نتيجة تيبس العضلات والمفاصل، لذى فقد يحتاج للتدخل الجراحي لتحرير الشد والتشوه حول المفصل ليعطي مجال أوسع لحرية الحركة، كما قد يحتاج الطفل للجراحة عند زيادة حدة تقوس العمود الفقري.
o القلب والأوعية الدموية: على الرغم من أن القلب. ليس مسبب رئيسي لمثل هذه الحالات، ولكن اقترح وجود علاقة بين أمراض القلبو مرض ضمور العضلات الشوكي بنوع ما.[22][23][24][25]
o الأدوية: هناك بعض الأدوية المستخدمة لعلاج الشد العضلي.
o الدعم النفسي والاجتماعي للطفل وعائلته.الأطفال المصابون لا يختلف سلوكهم عن عامة الناس؛ على الرغم من اصابتهم فان التطور المعرفي يمكن أن يكون أسرع قليلا، وفي بعض جوانب من الاستخبارات اشير إلى ان نسبة تعليمهم واستيعابهم أعلى من المتوسط.[26][27][28][29] ولكن الرعاية النفسية مهمة للتخفيفة عن المعاناة من المرض واساليب علاجه المؤلمة والمتعبة للمريض. بيان لتوافق مستوى الرعاية في ضمور العضلات الشوكي
العلاجات الناشئة
منذ وصفت الآلية الكامنة الجينية من SMA في عام 1990، تم اقتراح عدة طرق علاجية والتحقيق فيها. منذ عدد كبير من في المختبر ودراسات النمذجة الحيوانات تشير إلى أن استعادة مستويات SMN يعود أعراض SMA، فإن غالبية العلاجات الناشئة والتركيز على تعظيم توافر البروتين شبكة الإدارة العليا إلى الخلايا العصبية الحركية المسارات الرئيسية العلاجية قيد البحث اعتبارا من ديسمبر 2011.[30][31][32][33][34][35][36][37][38] وتتمثل في ما يلي:
العلاج الجيني - تجري حاليا أبحاثا باستخدام ناقلات فيروسية scAAV9 في جامعة ولاية أوهايو وعلى الصعيد الوطني مستشفى الأطفال، USA، وجامعة شيفيلد، المملكة المتحدة، فضلا عن شركة جنزايم، الولايات المتحدة الأمريكية، وجينيتون Généthon في فرنسا. في دراسة امكانية تصحيح وظيفة الجين إس إم أن1 من خلال إدراج النوكليوتيدات . وقد أدى هذه التجارب على الفئران في اطالة عمرها .[39]
العلاج بالخلايا الجذعية - يهدف إلى توفير الحماية للخلايا العصبية المتضررة من خلال حقن الشخص المصاب . أعدت خصيصا الخلايا الجذعية في النخاع الشوكي التي تنمي في وقت لاحق إلى خلايا العصبية قادرة إنتاج البروتين الداعم للجسم وخلاياه، تمت هذذه التجارب في الولايات المتحدة الأمريكية.و في العيادات الخاصة في البرازيل والصين وروسيا وأوكرانيا. وقد يطول الحديث حول النواحي الأخلاقية المتعلقة بعلاج النوع الشديد (النوع الأول) وهل يتم إجراء تنفس صناعي أم لا وهل ذلك في صالح الطفل ام يزيد ويطيل معاناته الجسدية والنفسية.
وصلات خارجية
- http://dx.doi.org/10.1177/0883073807305788 Standards of Care in Spinal Muscular Atrophy
- http://www.werathah.com/neuro/sma.htm
- http://www.amyotrophie-spinale.com
- http://www.myonet.org/
- http://www.amyotrophie-spinale.com
- http://www.myonet.org/
- http://www.doctissimo.fr/html/sante/encyclopedie/sa_996_mala_werdnig.htm
- http://www.doctissimo.fr/html/sante/encyclopedie/sa_996_mala_werdnig.htm
انظر أيضًا
المراجع
- Spinal muscular atrophy. - Abstract - Europe PMC نسخة محفوظة 2020-06-27 على موقع واي باك مشين.
- Spinal muscular atrophy. - Abstract - Europe PMC نسخة محفوظة 2020-06-27 على موقع واي باك مشين.
- OMIM Entry - # 253300 - SPINAL MUSCULAR ATROPHY, TYPE I; SMA1 نسخة محفوظة 18 يونيو 2017 على موقع واي باك مشين.
- OMIM Entry - # 253550 - SPINAL MUSCULAR ATROPHY, TYPE II; SMA2 نسخة محفوظة 04 مايو 2017 على موقع واي باك مشين.
- OMIM Entry - # 271150 - SPINAL MUSCULAR ATROPHY, TYPE IV; SMA4 نسخة محفوظة 10 مارس 2017 على موقع واي باك مشين.
- دُوِي:10.1016/S1090-3798(03)00060-6
- Krosschell، K. J.؛ Maczulski، J. A.؛ Crawford، T. O.؛ Scott، C.؛ Swoboda، K. J. (2006). "A modified Hammersmith functional motor scale for use in multi-center research on spinal muscular atrophy". Neuromuscular Disorders. ج. 16 ع. 7: 417–426. DOI:10.1016/j.nmd.2006.03.015. PMID:16750368.
- O'Hagen، J. M.؛ Glanzman، A. M.؛ McDermott، M. P.؛ Ryan، P. A.؛ Flickinger، J.؛ Quigley، J.؛ Riley، S.؛ Sanborn، E.؛ Irvine، C.؛ Martens، W. B.؛ Annis، C.؛ Tawil، R.؛ Oskoui، M.؛ Darras، B. T.؛ Finkel، R. S.؛ De Vivo، D. C. (2007). "An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients". Neuromuscular Disorders. ج. 17 ع. 9–10: 693–697. DOI:10.1016/j.nmd.2007.05.009. PMID:17658255.
- Glanzman، A. M.؛ O'Hagen، J. M.؛ McDermott، M. P.؛ Martens، W. B.؛ Flickinger، J.؛ Riley، S.؛ Quigley، J.؛ Montes، J.؛ Dunaway، S.؛ Deng، L.؛ Chung، W. K.؛ Tawil، R.؛ Darras، B. T.؛ De Vivo، D. C.؛ Kaufmann، P.؛ Finkel، R. S.؛ Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR) (2011). "Validation of the Expanded Hammersmith Functional Motor Scale in Spinal Muscular Atrophy Type II and III". Journal of Child Neurology. ج. 26 ع. 12: 1499–1507. DOI:10.1177/0883073811420294. PMID:21940700.
- قالب:SMN doi
- Jędrzejowska، M.؛ Milewski، M.؛ Zimowski، J.؛ Borkowska، J.؛ Kostera-Pruszczyk، A.؛ Sielska، D.؛ Jurek، M.؛ Hausmanowa-Petrusewicz، I. (2009). "Phenotype modifiers of spinal muscular atrophy: The number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease". Acta Biochimica Polonica. ج. 56 ع. 1: 103–108. PMID:19287802.
- Su، Y. N.؛ Hung، C. C.؛ Lin، S. Y.؛ Chen، F. Y.؛ Chern، J. P. S.؛ Tsai، C.؛ Chang، T. S.؛ Yang، C. C.؛ Li، H.؛ Ho، H. N.؛ Lee، C. N. (2011). Schrijver، Iris (المحرر). "Carrier Screening for Spinal Muscular Atrophy (SMA) in 107,611 Pregnant Women during the Period 2005–2009: A Prospective Population-Based Cohort Study". PLoS ONE. ج. 6 ع. 2: e17067. DOI:10.1371/journal.pone.0017067. PMC:3045421. PMID:21364876.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link) - Sugarman، E. A.؛ Nagan، N.؛ Zhu، H.؛ Akmaev، V. R.؛ Zhou، Z.؛ Rohlfs، E. M.؛ Flynn، K.؛ Hendrickson، B. C.؛ Scholl، T.؛ Sirko-Osadsa، D. A.؛ Allitto، B. A. (2011). "Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72 400 specimens". European Journal of Human Genetics. ج. 20 ع. 1: 27–32. DOI:10.1038/ejhg.2011.134. PMC:3234503. PMID:21811307.
- Little، S. E.؛ Janakiraman، V.؛ Kaimal، A.؛ Musci، T.؛ Ecker، J.؛ Caughey، A. B. (2010). "The cost-effectiveness of prenatal screening for spinal muscular atrophy". American Journal of Obstetrics and Gynecology. ج. 202 ع. 3: 253.2e1. DOI:10.1016/j.ajog.2010.01.032. PMID:20207244.
- Prior، T. W.؛ Professional Practice Guidelines Committee (2008). "Carrier screening for spinal muscular atrophy". Genetics in Medicine. ج. 10 ع. 11: 840–842. DOI:10.1097/GIM.0b013e318188d069. PMC:3110347. PMID:18941424.
- Rutkove، S. B.؛ Shefner، J. M.؛ Gregas، M.؛ Butler، H.؛ Caracciolo، J.؛ Lin، C.؛ Fogerson، P. M.؛ Mongiovi، P.؛ Darras، B. T. (2010). "Characterizing spinal muscular atrophy with electrical impedance myography". Muscle & Nerve. ج. 42 ع. 6: 915. DOI:10.1002/mus.21784.
{{استشهاد بدورية محكمة}}: الوسيط|إظهار المؤلفين=29غير صالح (مساعدة) - Messina، S.؛ Pane، M.؛ De Rose، P.؛ Vasta، I.؛ Sorleti، D.؛ Aloysius، A.؛ Sciarra، F.؛ Mangiola، F.؛ Kinali، M.؛ Bertini، E.؛ Mercuri، E. (2008). "Feeding problems and malnutrition in spinal muscular atrophy type II". Neuromuscular Disorders. ج. 18 ع. 5: 389–393. DOI:10.1016/j.nmd.2008.02.008. PMID:18420410.
- Chen، Y. S.؛ Shih، H. H.؛ Chen، T. H.؛ Kuo، C. H.؛ Jong، Y. J. (2011). "Prevalence and Risk Factors for Feeding and Swallowing Difficulties in Spinal Muscular Atrophy Types II and III". The Journal of Pediatrics. DOI:10.1016/j.jpeds.2011.08.016.
- دُوِي:10.1016/S1071-9091(98)80026-0
- Bach، J. R.؛ Niranjan، V.؛ Weaver، B. (2000). "Spinal Muscular Atrophy Type 1: A Noninvasive Respiratory Management Approach". Chest. ج. 117 ع. 4: 1100–1105. DOI:10.1378/chest.117.4.1100. PMID:10767247.
- Bach، J. R.؛ Saltstein، K.؛ Sinquee، D.؛ Weaver، B.؛ Komaroff، E. (2007). "Long-Term Survival in Werdnig–Hoffmann Disease". American Journal of Physical Medicine & Rehabilitation. ج. 86 ع. 5: 339. DOI:10.1097/PHM.0b013e31804a8505. PMID:17449977.
- Rudnik-Schoneborn، S.؛ Heller، R.؛ Berg، C.؛ Betzler، C.؛ Grimm، T.؛ Eggermann، T.؛ Eggermann، K.؛ Wirth، R.؛ Wirth، B.؛ Zerres، K. (2008). "Congenital heart disease is a feature of severe infantile spinal muscular atrophy". Journal of Medical Genetics. ج. 45 ع. 10: 635–638. DOI:10.1136/jmg.2008.057950. PMID:18662980.
- Heier، C. R.؛ Satta، R.؛ Lutz، C.؛ Didonato، C. J. (2010). "Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice". Human Molecular Genetics. ج. 19 ع. 20: 3906–3918. DOI:10.1093/hmg/ddq330. PMC:2947406. PMID:20693262.
- Shababi، M.؛ Habibi، J.؛ Yang، H. T.؛ Vale، S. M.؛ Sewell، W. A.؛ Lorson، C. L. (2010). "Cardiac defects contribute to the pathology of spinal muscular atrophy models". Human Molecular Genetics. ج. 19 ع. 20: 4059–4071. DOI:10.1093/hmg/ddq329. PMID:20696672.
- Bevan، A. K.؛ Hutchinson، K. R.؛ Foust، K. D.؛ Braun، L.؛ McGovern، V. L.؛ Schmelzer، L.؛ Ward، J. G.؛ Petruska، J. C.؛ Lucchesi، P. A.؛ Burghes، A. H. M.؛ Kaspar، B. K. (2010). "Early heart failure in the SMNΔ7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery". Human Molecular Genetics. ج. 19 ع. 20: 3895–3905. DOI:10.1093/hmg/ddq300. PMC:2947399. PMID:20639395.
- دُوِي:10.1016/S0960-8966(01)00274-7
- دُوِي:10.1016/S0960-8966(06)80008-8
- Laufersweiler-Plass، C.؛ Rudnik-Schöneborn، S.؛ Zerres، K.؛ Backes، M.؛ Lehmkuhl، G.؛ Von Gontard، A. (2002). "Behavioural problems in children and adolescents with spinal muscular atrophy and their siblings". Developmental Medicine & Child Neurology. ج. 45. DOI:10.1017/S0012162203000082.
- De Oliveira، C. M.؛ Araújo، A. P. D. Q. C. (2011). "Self-reported quality of life has no correlation with functional status in children and adolescents with spinal muscular atrophy". European Journal of Paediatric Neurology. ج. 15 ع. 1: 36–39. DOI:10.1016/j.ejpn.2010.07.003. PMID:20800519.
- Pruss، R. M.؛ Giraudon-Paoli، M.؛ Morozova، S.؛ Berna، P.؛ Abitbol، J. L.؛ Bordet، T. (2010). "Drug discovery and development for spinal muscular atrophy: Lessons from screening approaches and future challenges for clinical development". Future Medicinal Chemistry. ج. 2 ع. 9: 1429–1440. DOI:10.4155/FMC.10.228. PMID:21426138.
- Sproule، D. M.؛ Kaufmann، P. (2010). "Therapeutic developments in spinal muscular atrophy". Therapeutic Advances in Neurological Disorders. ج. 3 ع. 3: 173–185. DOI:10.1177/1756285610369026. PMC:3002649. PMID:21179609.
- Fuller، H. R.؛ Barišić، M.؛ Šešo-Šimić، Đ. I.؛ Špeljko، T.؛ Morris، G. E.؛ Šimić، G. (2010). "Treatment strategies for spinal muscular atrophy". Translational Neuroscience. ج. 1 ع. 4: 308. DOI:10.2478/v10134-010-0045-4.
- Sendtner، M. (2010). "Therapy development in spinal muscular atrophy". Nature Neuroscience. ج. 13 ع. 7: 795–799. DOI:10.1038/nn.2565. PMID:20581815.
- Bosboom، W. M.؛ Vrancken، A. F. E.؛ Van Den Berg، L. H.؛ Wokke، J. H.؛ Iannaccone، S. T. (2009). Bosboom، Wendy MJ (المحرر). "Cochrane Database of Systematic Reviews". DOI:10.1002/14651858.CD006281.pub2.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) والوسيط|الفصل=تم تجاهله (مساعدة) - Bosboom، W. M.؛ Vrancken، A. F. E.؛ Van Den Berg، L. H.؛ Wokke، J. H.؛ Iannaccone، S. T. (2009). Bosboom، Wendy MJ (المحرر). "Cochrane Database of Systematic Reviews". DOI:10.1002/14651858.CD006282.pub2.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) والوسيط|الفصل=تم تجاهله (مساعدة) - Wadman، R. I.؛ Bosboom، W. M.؛ Van Den Berg، L. H.؛ Wokke، J. H.؛ Iannaccone، S. T.؛ Vrancken، A. F. E. (2011). Wadman، Renske I (المحرر). "Cochrane Database of Systematic Reviews". DOI:10.1002/14651858.CD006281.pub3.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) والوسيط|الفصل=تم تجاهله (مساعدة) - Wadman، R. I.؛ Bosboom، W. M.؛ Van Den Berg، L. H.؛ Wokke، J. H.؛ Iannaccone، S. T.؛ Vrancken، A. F. E. (2011). Wadman، Renske I (المحرر). "Cochrane Database of Systematic Reviews". DOI:10.1002/14651858.CD006282.pub3.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) والوسيط|الفصل=تم تجاهله (مساعدة) - Lewelt، A.؛ Newcomb، T. M.؛ Swoboda، K. J. (2011). "New Therapeutic Approaches to Spinal Muscular Atrophy". Current Neurology and Neuroscience Reports. ج. 12 ع. 1: 42–53. DOI:10.1007/s11910-011-0240-9. PMC:3260050. PMID:22134788.
- Bevan، A. K.؛ Duque، S.؛ Foust، K. D.؛ Morales، P. R.؛ Braun، L.؛ Schmelzer، L.؛ Chan، C. M.؛ McCrate، M.؛ Chicoine، L. G.؛ Coley، B. D.؛ Porensky، P. N.؛ Kolb، S. J.؛ Mendell، J. R.؛ Burghes، A. H.؛ Kaspar، B. K. (2011). "Systemic Gene Delivery in Large Species for Targeting Spinal Cord, Brain, and Peripheral Tissues for Pediatric Disorders". Molecular Therapy. ج. 19 ع. 11: 1971–1980. DOI:10.1038/mt.2011.157. PMC:3222525. PMID:21811247.
 بوابة طب
بوابة طب