سوء التغذية العضلي
عسر النمو العَضلي[1][2][3][4] أو الحثل العضلي[5] أو سوء التغذية العضلي[5] أو فساد عضلي وهو اضطراب جيني نادر، يتقمّص عدّة مظاهر؛ منه الكلّي الخطير (حثل دِوشين دو بولونْيْ)، يصيب الذّكور فقط ويترك الإناث، يبدؤ مبكّرا في الطّفولة بإصابة الحزامين الحوضي (مشية مترنّحة وصعوبة القيام بعد الجلوس وصعوبة ارتقاء السّلالم..) والكتفي ثمّ تنتقل الإصابة إلى الجذع فالوجه فعامّة العضلات (محدثا تشوّهات) حتّى في القلب والتّنفّس مميتان، وهناك إصابات حِثلية أخرى عديدة متأخرة وبطيئة التّطوّر وغير خطيرة تسمح بحياة عادية تقريبا.
| سوء التغذية العضلي | |
|---|---|
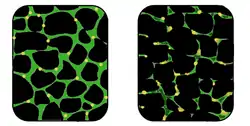 في العضلة المصابة (إلى اليمين) يصبح النسيج غير منتظم ويقل تركيز الديستروفين (بالأخضر) بشكل كبير. مقارنة مع العضلة الطبيعية (إلى لليسار). في العضلة المصابة (إلى اليمين) يصبح النسيج غير منتظم ويقل تركيز الديستروفين (بالأخضر) بشكل كبير. مقارنة مع العضلة الطبيعية (إلى لليسار). | |
| معلومات عامة | |
| الاختصاص | طب الجهاز العصبي، وطب الأطفال، وعلم الوراثة الطبية |
| من أنواع | اعتلال عضلي، ومرض |
الأسباب
الدستروفين هذه الحالات موروثة عمومًا، وتتبع الضمور العضلي أنماط الوراثة المختلفة. يمكن أن يرث الأفراد الحثل العضلي كاضطراب مرتبط بالأكسجين أو اضطراب متنحي أو مهيمن. علاوة على ذلك، يمكن أن يكون طفرة عفوية مما يعني وجود أخطاء في تكرار الحمض النووي والآفات العفوية. الآفات العفوية ناتجة عن أضرار طبيعية للحمض النووي، حيث الأكثر شيوعًا هي إزالة الرطوبة.[6][7]
تم العثور على بروتين الدستروفين في غشاء ليف العضلات. طبيعتها الحلزونية تسمح لها بالتصرف مثل نابض أو ممتص صدمات. يربط الدستروفين الأكتين في الهيكل الخلوي والدستروجليكان في غشاء بلازما الخلايا العضلية، المعروف باسم الساركوليما (خارج الخلية). بالإضافة إلى التثبيت الميكانيكي، ينظم الديستروفين أيضًا مستويات الكالسيوم.[8][9] يقع جين ديستروفين على كروموسوم إكس. في الذكور، يحتوي كروموسوم لون X على جين واحد فقط من ديستروفين. إذا كان هناك طفرة في هذا الجين، فستفتقد عضلات الذكر للديستروفين وتتحلل ببطء؛ تم تحديد طفرات في الجين الديستروفين كسبب لـ حثل العضلى من قبل باحثين رابطة الضمور العضلي في عام 1986. لدى الأنثى دائمًا جينات الديستروفين واحدة على كل كروموسوم X، وحتى إذا كان أحد هذه الجينات لا يعمل، فإن الجين الآخر يكفي للحفاظ على مستويات الديستروفين عالية بما فيه الكفاية للحفاظ على وظيفة العضلات في كل من عضلات القلب والهيكل العظمي. ومع ذلك، فقد أظهرت الأبحاث أن أقلية صغيرة من الإناث اللاتي لديهن جين يعمل الديستروفين العامل وغير العامل يمكن أن تظهر عليه أعراض حثل عضلى أظهرت الدراسات الحديثة على تفاعل البروتينات مع طفرات خاطئة وجيرانها درجة عالية من الصلابة. يرتبط بالبروتينات المركزية المركزية المشاركة في ربط البروتينات والشبكات الفرعية المرنة ذات الوظائف الجزيئية المرتبطة بالكالسيوم.[10]
العلاج
ليس الحثل العضلي الوراثي علاج عقاري فعّال، وإنّما يتطلّب ترويضا طبّيا خاصّا لتحسين حالة المريض الحركية وتدخّلات جراحية لتقويم التّشوّهات واحتياطات حتّى لا تُرهَق العضلات (إذ ليس معنى عدم القدرة على تسلّق السّلالم.. قلّة التّدريب، ومن الاحتياطات الواجب اتباعها زواج الفتيات حاملة الصفة الوراثية إجراء فحص للجنين في بداية الحمل قبل تخليق الجنين أو إجراء التخصيب المخبرى «أطفال الانابيب» للاجنة السليمة -والتأكد من عدم انتقال الصفة الوراثية وبالتالى انتقال المرض إلى الأولاد- واجهاض الجنين قبل تخليقه،ومن الجدير بالذكر ان الذكر المصاب بالمرض لا ينقل الصفة الوراثية لأبناءه في حالة زواجه من سليمة. اى ان الانثي حاملة الصفة تنقل الصفة رغم انها لا تظهر عليها اعراض المرض بتاتا اماالذكر وان كان مصابا وظاهره عليه اعراض المرض فانه لا ينقل الصفة.حيث ان الصفة تحمل على الكروموسوم إكس.
تاريخيا
في عام 1860، أصبحت أوصاف الأولاد الذين أصبحوا أضعف تدريجيًا وفقدوا القدرة على المشي وتوفيوا في سن مبكرة أكثر بروزًا في المجلات الطبية. في العقد التالي،[11] لقدم عالم الأعصاب الفرنسي غيوم دوشين وصفًا شاملاً لأكثر أشكال المرض شيوعًا وشدة، والذي يحمل اسمه الآن الحثل العضلي الدوشيني
التصنيف
| الوراثة المندلية عبر الإنترنت | الجين | الوصف | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| حثل بيكر العضلي | 300376 | DMD | حثل بيكر العضلي (BMD) هو نوع أقل شدة من حثل دوشين العضلي وينتج عن إنتاج شكل حثل مقطوع، لكن وظيفي جزئيًا عن الدستروفين
[12] البقاء على قيد الحياة عادة ما يكون في سن الشيخوخة ويصيب الأولاد فقط (مع استثناءات نادرة للغاية[13] | |||||||
| حثل عضلي خلقي | Multiple | Multiple | .jpg.webp) Hydrocephalus |
العمر عند البدء هو الولادة، وتشمل الأعراض ضعف العضلات العام وتشوهات المفاصل المحتملة، والمرض يتقدم ببطء، ويقصر عمر المريض. يتضمن حثل العضلي الخلقي العديد من الاضطرابات مع مجموعة من الأعراض. قد يكون تنكس العضلات خفيفًا أو شديدًا. قد تقتصر المشكلات على العضلات والهيكل العظمي، أو يمكن أن يقترن تنكس العضلات بالتأثيرات على المخ والأنظمة الأخرى للأعضاء.[14]
تنجم عدة أشكال من حثل العضلات الخلقية عن عيوب في البروتينات التي يعتقد أن لها علاقة ما بمركب بروتين الدستروفين السكري والصلات بين الخلايا العضلية وهيكلها الخلوي المحيط بها. تظهر بعض أشكال حثل العضلي الخلقي تشوهات دماغية حادة، مثل السعال واستسقاء الرأس.[15] يتراوح العمر الافتراضي بين 15 و 45، على الرغم من وجود استثناءات قليلة. حدد الباحثون الجين الذي يصيب ديستروفين البروتين، والذي، عندما يتغيب، يسبب حثل دوتشين[16] نظرًا لأن الجين موجود على كروموسوم X، فإن هذا الاضطراب يصيب الذكور في المقام الأول، والإناث اللائي يحملن أعراض أكثر اعتدالًا. تحدث طفرات متفرقة في هذا الجين بشكل متكرر.[17] دستروفين هو جزء من بنية معقدة تنطوي على العديد من مكونات البروتين الأخرى. يساعد "مجمع بروتين الدوستفرين " في ترسيخ الهيكل العظمي الهيكلي (الهيكل الخلوي) داخل خلايا العضلات، من خلال الغشاء الخارجي (sarcolemma) لكل خلية، إلى إطار الأنسجة (المصفوفة خارج الخلية) التي تحيط بكل خلية. بسبب العيوب في هذا التجمع، يؤدي تقلص العضلات إلى تعطيل الغشاء الخارجي للخلايا العضلية وإضعاف العضلات وإضاعةها في نهاية المطاف.[18] اعتلال ميوشي، أحد حَثَلُ العَضَلاَتِ القاصِيَة، يسبب ضعفًا أوليًا في عضلات ربلة الساق، ويتسبب عن عيوب في نفس الجين المسؤول عن أحد أشكال حثل عضلات الأطراف الحركية.[19][20] الأنواع الفرعية الثلاثة لإيمري-دريفوس قابلة للتمييز حسب نمط الميراث الخاص بهم:.X- المترابط، التلقائي المهيمن، والمتنحي التلقائي النموذج X- المرتبط هو الأكثر شيوعًا. كل نوع يختلف في انتشار والأعراض. يحدث المرض بسبب طفرات في جين LMNA، أو أكثر شيوعًا، في جين EMD. كلا الجينات ترميز لمكونات البروتين من المغلف النووي. ومع ذلك، كيف تسبب هذه الطفرات التسبب في المرض ليست مفهومة جيدا.[21] | ||||||
| 158900 | DUX4 |
الحَثَلُ العَضَلِيُّ الوَجْهِيُّ الكَتِفِيُّ العَضُدِيُّ العضلات في الوجه والكتفين والذراعين العلويين مع الضعف التدريجي.[22] تتطور الاعراض عاده في مرحله البلوغ المبكر (المراهقون المتاخرون) ؛ يصاب الافراد المتضررون باعاقه شديده. نمط الميراث هو المهيمن التلقائي، علي الرغم من حدوث عدد من الطفرات العفوية. هناك حاجه إلى اثنين من العيوب لل FHHD، والتي توفر للمرة الاولي نظرية موحده لعلم الوراثة الاساسيه.[12][23] تحدث المراه البشرية في الذكور والإناث. [24] |
حثل التأتر العضل | 160900, 602668 | DMPK, CNBP
حثل العضلي العضلي هو حالة سائدة وراثيّة تظهر مع توتر العضلات (تأخر استرخاء العضلات)، إضافة إلى هزال العضلات وضعفها.[25] يختلف حَثَلُ التَّأَتُّرِ العَضَلِيّ في شدته ومظاهره ويؤثر على العديد من أجهزة الجسم بالإضافة إلى عضلات الهيكل العظمي، بما في ذلك القلب وأعضاء الغدد الصماء والعينين.[26] تظهر جميع حَثَلُ عَضَلاَتِ حِزامِ الطَّرَف توزيعا مماثلا لضعف العضلات، مما يؤثر على كل من الذراعين والساقين. تم التعرف على العديد من أشكاله، والتي تظهر أنماطًا مختلفة من الميراث (جسمية مقهورة مقابل جسمية جسمية). في نمط راثي متنحي من الميراث، يتلقى الفرد نسختين من الجين المعيب، واحدة من كل والد. المتنحية هي أكثر تواترا من الأشكال السائدة، وعادة ما يكون ظهور الطفولة أو المراهقة. حَثَلُ عَضَلاَتِ حِزامِ الطَّرَف المهيمنة عادة ما تظهر بداية الكبار. ارتبطت بعض الأشكال المتنحية بعيوب في البروتينات التي تشكل مجمع بروتين الدستروفين.[27] |
الحثل العيني البلعومي | 164300 | PABPN1 | الحَثَلُ العَينِيُّ البُلْعومِي ّالعمر في بداية 40 إلى 70 سنة؛ تؤثر الأعراض على عضلات الجفون والوجه والحلق تليها ضعف عضلات الحوض والكتف. ويعزى ذلك إلى توسع قصير في الجينوم الذي ينظم ترجمة بعض الجينات إلى بروتينات وظيفية.[12] |
|-
إداريا
حاليا، لا يوجد علاج لضمور العضلات. من حيث الاداره، والعلاج الطبيعي، والعلاج الوظيفي، والتدخل تقويم العظام (على سبيل المثال، الكاحل القدم العظام) ،[28][29] العلاج بالكلام، والعلاج التنفسي قد يكون مفيدا[28] القشرية منخفضه الكثافة مثل البريدنيزون، وقد يساعد الانكماش العضلي علي الحفاظ علي تناغم العضلات.[30] قد تكون هناك حاجه إلى الاجهزه التقويمية (أدوات تقويم العظام المستخدمة للدعم) وجراحه تقويم العظام التصحيحية لتحسين نوعيه الحياة في بعض الحالات.[31] قد تتطلب المشاكل القلبية التي تحدث مع الضمور العضلي الذي يحدث مع EDMD وجهاز تنظيم ضربات القلب.[32] ويمكن علاج ميوفونيا (تاخر الاسترخاء في العضلات بعد انكماش قوي) الذي يحدث في ضمور العضلات العضلية مع الادويه مثل الكينين.[33]
العلاج المهني يساعد الفرد مع الحثل العضلى للانخراط في أنشطه الحياة اليومية (مثل التغذية الذاتية وأنشطه الرعاية الذاتية) والانشطه الترفيهية علي المستوي الأكثر استقلالا ممكن. ويمكن تحقيق ذلك باستخدام المعدات التكيفيه أو استخدام تقنيات حفظ الطاقة. وقد يؤدي العلاج المهني إلى إدخال تغييرات علي بيئة الشخص، سواء في المنزل أو العمل، لزيادة وظيفة الفرد وامكانيه الوصول اليه؛ وعلاوة علي ذلك، فانه يتناول التغييرات النفسانية والاجتماعية والتدهور المعرفي التي قد تصاحب MD، ويوفر الدعم والتثقيف حول هذا المرض للاسره والفرد.[34]
توقعات سير المرض
توقعات سير المرض يعتمد على شكل فردي من الحثل العضلى. في بعض الحالات، سصبح الشخص المصاب بمرض عضلي أضعف تدريجيًا إلى الحد الذي يؤدي إلى تقصير عمر المريض بسبب مضاعفات القلب والتنفس. ومع ذلك، فإن بعض أمراض العضلات لا تؤثر على متوسط العمر المتوقع على الإطلاق، وتحاول الأبحاث الجارية العثور على علاجات وعلاجات لإبطاء ضعف العضلات.[31]
بحثيا
أجرت منظمة الصحة العالمية تجارب على نظام الستيرويد الأمثل للدواء، في المملكة المتحدة في عام 2012.[35] فيما يتعلق بالبحوث داخل الولايات المتحدة، فإن المؤسسات الرئيسية الممولة من الحكومة الفيدرالية والتي تركز على أبحاث الضمور العضلي، بما في ذلك العلاج الجيني والطب التجديدي، هي المعهد الوطني للاضطرابات العصبية والسكتة الدماغية، والمعهد الوطني لالتهاب المفاصل وأمراض العضلات والعظام والجلد، المعهد الوطنى لصحة الطفل والتنمية.[12] في عام 1966، بدأت رابطة الحثل العضلي التابعه لجيرى لويس السنوي، والذي ربما يكون قد بذل المزيد من الجهد لرفع الوعي حول حثل العضلات أكثر من أي حدث أو مبادرة أخرى. ومع ذلك، فقد انتقد المدافعون عن حقوق الإعاقة التيليثون لتصوير ضحايا المرض على أنهم يستحقون الشفقة وليس الاحترام.[36] في 18 ديسمبر 2001، تم توقيع قانون لرعاية مرضى حثل العضلات في الولايات المتحدة الأمريكية. يعدل قانون خدمات الصحة العامة لتوفير البحوث لمختلف الحثل العضلي. أنشأ هذا القانون أيضًا لجنة تنسيق الحثل العضلي للمساعدة في تركيز الجهود البحثية من خلال إستراتيجية بحثية متماسكة.[37][38]
انظر أيضًا
روابط خارجية
- Muscular dystrophies على مشروع الدليل المفتوح
- National Registry for Myotonic Dystrophy and FSHD
- FSH Society for Fascioscapulohumeral muscular dystrophy (USA and Worldwide)
- FSHD Canada Foundation (CAN)
- FSHD Europe (EU)
- FSHD Global Research Foundation (Australia)
- FSHD Stichting (Netherlands)
مراجع
- كتاب الأحياء للصف الثاني ثانوي العلمي، طبعة عام 1997،صفحة 205، المملكة الأردنية الهاشمية.
- Finger, Stanley; Boller, Francois; Tyler, Kenneth L. (8 Dec 2009). History of Neurology: Handbook of Clinical Neurology (Series Editors: Aminoff, Boller and Swaab) (بالإنجليزية). Elsevier. p. 477. ISBN:9780702035418. Archived from the original on 2017-01-06.
- "Duchenne Muscular Dystrophy. What is muscular dystrophy? | Patient". Patient.info. 15 أبريل 2016. مؤرشف من الأصل في 2016-12-02. اطلع عليه بتاريخ 2017-03-14.
- Jenkins، Simon P.R. (2005). Sports Science Handbook:I - Z. Brentwood, Essex: Multi-Science Publ. Co. ص. 121. ISBN:0906522-37-4.
- المعجم الطبي الموحد. نسخة محفوظة 26 سبتمبر 2017 على موقع واي باك مشين.
- Choices، NHS. "Muscular dystrophy - Causes - NHS Choices". www.nhs.uk. مؤرشف من الأصل في 2016-04-02. اطلع عليه بتاريخ 2016-04-10.
- Griffiths, Anthony JF; Miller, Jeffrey H.; Suzuki, David T.; Lewontin, Richard C.; Gelbart, William M. (1 Jan 2000). "Spontaneous mutations" (بالإنجليزية). Archived from the original on 2017-04-02.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(help) - "DMD gene". Genetics Home Reference. 28 مارس 2016. مؤرشف من الأصل في 2016-04-16. اطلع عليه بتاريخ 2016-04-10.
- Lapidos, Karen A.; Kakkar, Rahul; McNally, Elizabeth M. (30 Apr 2004). "The Dystrophin Glycoprotein Complex Signaling Strength and Integrity for the Sarcolemma". Circulation Research (بالإنجليزية). 94 (8): 1023–1031. DOI:10.1161/01.RES.0000126574.61061.25. ISSN:0009-7330. PMID:15117830. Archived from the original on 2016-05-25.
- Sharma، Ankush (2014). "Publication:Rigidity and flexibility in protein-protein interaction networks: a case study on neuromuscular disorders". www.openaire.eu. articleId=od________18%3A%3A4d9ab382601b71369f95cfeeaf9ace55 مؤرشف من الأصل في 2016-04-22. اطلع عليه بتاريخ 2016-04-10.
{{استشهاد بدورية محكمة}}: تحقق من قيمة|مسار أرشيف=(مساعدة) - Laing، Nigel G؛ Davis، Mark R؛ Bayley، Klair؛ Fletcher، Sue؛ Wilton، Steve D (1 أغسطس 2011). "Molecular Diagnosis of Duchenne Muscular Dystrophy: Past, Present and Future in Relation to Implementing Therapies". The Clinical Biochemist Reviews. ج. 32 ع. 3: 129–134. ISSN:0159-8090. PMC:3157948. PMID:21912442.
- May 2006 report to Congress نسخة محفوظة 2014-04-05 على موقع واي باك مشين. on Implementation of the MD CARE Act, as submitted by Department of Health ad Human Service's معاهد الصحة الوطنية الأمريكية
- "Becker muscular dystrophy: MedlinePlus Medical Encyclopedia". medlineplus.gov (بالإنجليزية). Archived from the original on 2017-03-15. Retrieved 2017-03-14.
- Congenital Muscular Dystrophy~clinical في موقع إي ميديسين
- |- |الحثل العضلى دوشين |310200 |DMD |- |الحثل العضلي دوتشين هو الشكل الأكثر شيوعًا في الطفولة من الحثل العضلي. يصيب بشكل عام الأولاد فقط (مع استثناءات نادرة للغاية)، ويصبح واضحًا سريريًا عندما يبدأ الطفل في المشي. بحلول سن العاشرة، قد يحتاج الطفل إلى حمالات للمشي وبحلول سن الثانية عشرة، يكون معظم المرضى غير قادرين على المشي.<ref name="MedlinePlusEncyclopedia000705">"Duchenne muscular dystrophy: MedlinePlus Medical Encyclopedia". medlineplus.gov (بالإنجليزية). Archived from the original on 2017-04-05. Retrieved 2017-03-14.
- "Duchenne and Becker muscular dystrophy - Genetics Home Reference". Ghr.nlm.nih.gov. 7 مارس 2017. مؤرشف من الأصل في 2017-03-24. اطلع عليه بتاريخ 2017-03-14.
- "Duchenne Muscular Dystrophy. What is muscular dystrophy? | Patient". Patient.info. 15 أبريل 2016. مؤرشف من الأصل في 2016-12-02. اطلع عليه بتاريخ 2017-03-14.
-
|-
|حثل العضلات القاصية
|254130
|DYSF
|
يتراوح عمر حَثَلُ العَضَلاَتِ القاصِيَة عند بداية المرض من 20 إلى 60 عامًا تقريبًا ؛ تشمل الأعراض ضعف وإضاعة عضلات اليدين والساعدين والساقين السفلية ؛ التقدم بطيء ولا يهدد الحياة.
<ref>Udd، Bjarne (2011). Distal muscular dystrophies. ج. 101. ص. 239–62. DOI:10.1016/B978-0-08-045031-5.00016-5. ISBN:9780080450315. PMID:21496636.
{{استشهاد بكتاب}}:|صحيفة=تُجوهل (مساعدة) - |- |إيمري-دريفوس حثل العضلي |310300, 181350 |EMD, LMNA |إيمري دريفوس حثل العضليمرضى حثل العضلي يتواجدون عادة في مرحلة الطفولة وسنوات المراهقة المبكرة مع تقلصات. تشمل العلامات السريرية ضعف العضلات وهزالها، بدءًا من عضلات الأطراف البعيدة والتقدم لإشراك عضلات أطرافهم. يعاني معظم المرضى أيضًا من عيوب التوصيل القلبي وعدم انتظام ضربات القلب. Emery–Dreifuss muscular dystrophy patients normally present in childhood and the early teenaged years with contractures. Clinical signs include muscle weakness and wasting, starting in the distal limb muscles and progressing to involve the limb-girdle muscles. Most patients also suffer from cardiac conduction defects and arrhythmias.<ref>"OMIM Entry - # 310300 - EMERY-DREIFUSS MUSCULAR DYSTROPHY 1, X-LINKED; EDMD1". Omim.org. مؤرشف من الأصل في 2017-03-10. اطلع عليه بتاريخ 2017-03-14.
- "Emery-Dreifuss muscular dystrophy - Genetics Home Reference". Ghr.nlm.nih.gov. 7 مارس 2017. مؤرشف من الأصل في 2017-03-12. اطلع عليه بتاريخ 2017-03-14.
- Emery–Dreifuss Muscular Dystrophy في موقع إي ميديسين
- "facioscapulohumeral muscular dystrophy - Genetics Home Reference". Ghr.nlm.nih.gov. مؤرشف من الأصل في 2017-03-24. اطلع عليه بتاريخ 2017-03-14.
- Rosenberg، Roger N.؛ Pascual، Juan M. (28 أكتوبر 2014). Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease. ص. 1174. ISBN:978-0124105492. مؤرشف من الأصل في 2017-03-15. اطلع عليه بتاريخ 2017-03-14.
- "Facioscapulohumeral muscular dystrophy: MedlinePlus Medical Encyclopedia". Nlm.nih.gov. 9 مارس 2017. مؤرشف من الأصل في 2016-07-04. اطلع عليه بتاريخ 2017-03-14.
- Turner، C.؛ Hilton-Jones، D. (2010). "The myotonic dystrophies: diagnosis and management" (PDF). Journal of Neurology, Neurosurgery & Psychiatry. ج. 81 ع. 4: 358–67. DOI:10.1136/jnnp.2008.158261. PMID:20176601. مؤرشف من الأصل (PDF) في 2019-10-23.
- "Myotonic Dystrophy Type 1". Myotonic Dystrophy Type 1 - GeneReviews® - NCBI Bookshelf. University of Washington, Seattle. 1993. مؤرشف من الأصل في 2017-01-18. اطلع عليه بتاريخ 2017-03-14.
{{استشهاد بكتاب}}:|موقع=تُجوهل (مساعدة)</ref حَثَلُ التَّأَتُّرِ العَضَلِيّ الاول هو الشكل الأكثر شيوعًا لدى البالغين. وينتج عن تمدد تكرار قصير في تسلسل الحمض النووي لجين كيناز بروتين الحثل العضلي. حثل العضلات العضلي الثانى نادر الحدوث، وهو نتيجة لتوسيع CCTG في جين بروتين إصبع الزنك.<ref name="2006 report to Congress" /> |- |حثل عضلات حزام الطرف |158900 |DUX4 | | حَثَلُ عَضَلاَتِ حِزامِ الطَّرَف يؤثر على كل من الفتيان والفتيات.<ref>Pegoraro، E؛ Hoffman، EP؛ Adam، MP؛ Ardinger، HH؛ Pagon، RA؛ Wallace، SE؛ Bean، LJH؛ Stephens، K؛ Amemiya، A (2012). "Limb-Girdle Muscular Dystrophy Overview". PMID:20301582.{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) - على الرغم من أن الشخص عادة ما يعيش حياة طبيعية مع بعض المساعدة، إلا أنه في بعض الحالات القصوى، تحدث الوفاة الناجمة عن إل جي إم دي بسبب مضاعفات القلب والأوعية الدموية.<ref>Jenkins، Simon P.R. (2005). Sports Science Handbook:I - Z. Brentwood, Essex: Multi-Science Publ. Co. ص. 121. ISBN:978-0906522-37-0.
- "What are the treatments for muscular dystrophy?". NIH.gov. NIH. 2015. مؤرشف من الأصل في 2016-04-07. اطلع عليه بتاريخ 2016-04-10.
- "Muscular Dystrophy-OrthoInfo - AAOS". orthoinfo.aaos.org. مؤرشف من الأصل في 2016-04-12. اطلع عليه بتاريخ 2016-04-10.
- McADAM، LAURA C.؛ MAYO، AMANDA L.؛ ALMAN، BENJAMIN A.؛ BIGGAR، W. DOUGLAS (1 مايو 2012). "The Canadian experience with long term deflazacort treatment in Duchenne muscular dystrophy". Acta Myologica. ج. 31 ع. 1: 16–20. ISSN:1128-2460. PMC:3440807. PMID:22655512.
- "Muscular Dystrophy: Hope Through Research". NINDS. 4 مارس 2016. مؤرشف من الأصل في 2016-09-30. اطلع عليه بتاريخ 2016-09-12.
- Verhaert، David؛ Richards، Kathryn؛ Rafael-Fortney، Jill A.؛ Raman، Subha V. (1 يناير 2011). "Cardiac Involvement in Patients with Muscular Dystrophies: Magnetic Resonance Imaging Phenotype and Genotypic Considerations". Circulation: Cardiovascular Imaging. ج. 4 ع. 1: 67–76. DOI:10.1161/CIRCIMAGING.110.960740. ISSN:1941-9651. PMC:3057042. PMID:21245364.
- Eddy, Linda L. (25 Jan 2013). Caring for Children with Special Healthcare Needs and Their Families: A Handbook for Healthcare Professionals (بالإنجليزية). John Wiley & Sons. ISBN:9781118517970. Archived from the original on 2017-01-06.
- Lehman، R. M.؛ McCormack، G. L. (2001). "Neurogenic and Myopathic Dysfunction". في Pedretti، Lorraine Williams؛ Early، Mary Beth (المحررون). Occupational Therapy: Practice Skills for Physical Dysfunction (ط. 5th). Mosby. ص. 802–3. ISBN:978-0-323-00765-8.
- Choices، N. H. S. (9 نوفمبر 2011). "Muscular Dystrophy - Clinical trial details - NHS Choices". مؤرشف من الأصل في 2016-04-21. اطلع عليه بتاريخ 2016-04-10.
- Berman، Ari (2 سبتمبر 2011). "The End of the Jerry Lewis Telethon—It's About Time". The Nation. مؤرشف من الأصل في 2015-04-18. اطلع عليه بتاريخ 2017-03-14.
- H.R. 717--107th Congress (2001) نسخة محفوظة 2012-02-19 على موقع واي باك مشين.: MD-CARE Act, GovTrack.us (database of federal legislation), (accessed Jul 29, 2007)
- Public Law 107-84 نسخة محفوظة 2012-11-07 على موقع واي باك مشين., PDF as retrieved from معاهد الصحة الوطنية الأمريكية website [وصلة مكسورة]
 بوابة طب
بوابة طب