خلل التنسج المشاشي المتعدد
مرض فيربانك أو خلل التنسج المشاشي المتعدد (MED) هو اضطراب وراثي نادر (الشكل السائد: حالة من كل 10000 ولادة) يصيب الأطراف النامية للعظام. تتطاول العظام الطويلة عادةً عن طريق توسع الغضاريف ضمن صفيحة النمو (الصفيحة المشاشية) بالقرب من نهاياتها. أثناء امتداد الغضروف خارج صفيحة النمو، يتمعدن وتزداد صلابته ليصبح عظمًا (التعظم). في MED، تتعطل هذه العملية.
| خلل التنسج المشاشي المتعدد | |
|---|---|
| Multiple epiphyseal dysplasia | |
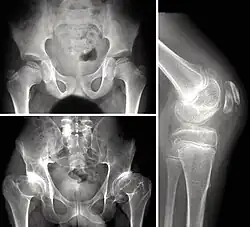 أشعة سينية لطفل يعاني من خلل التنسج المشاشي المتعدد أشعة سينية لطفل يعاني من خلل التنسج المشاشي المتعدد | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | خلل تنسج عظمي غضروفي |
| المظهر السريري | |
| الأعراض | ألم مفصلي[1] |
العلامات والأعراض
يعاني الأطفال المصابون بـMED -الذي ينتقل بصفة جسدية سائدة- من ألم في المفاصل وتعب بعد ممارسة الرياضة. يظهر التصوير بالأشعة السينية مراكز تعظم صغيرة وغير منتظمة، أكثرها وضوحًا في الوركين والركبتين. توجد العديد من مشاشات رأس الفخذ الصغيرة جدًا، بالإضافة لأسقف أجواف حقية ناقصة التنسج وضعيفة التشكل.[2] قد تتطور مِشية متهادية. يكون للركبتين اتساع كردوسي وخلل بينما تكون أصابع الأيدي قصيرة وعظام السنع مدورة ومتقاربة. القدم المسطحة شائعة جدًا.[3] العمود الفقري طبيعي لكن قد يكون فيه بعض العيوب، مثل الجنف.
عند البلوغ، يكون المصابون قصيري القامة أو في الحد الأدنى الطبيعي من الطول ويملكون أطرافًا قصيرة مقارنةً بجذوعهم. في كثير من الأحيان، تصبح حركة المفاصل الرئيسية محدودة، وخاصة في المرفقين والورك. ومع ذلك، يمكن أن تحدث ليونة في مفصل الركبة والأصابع. تبدأ علامات الفصال العظمي عادةً مبكرًا في مرحلة البلوغ.[4]
يعاني الأطفال المصابون بـ MED كصفة متنحية من آلام في المفاصل، خاصةً في الورك والركبتين، وعادةً ما يعانون من تشوهات في اليدين والقدمين والركبتين أو العمود الفقري (مثل الجنف). يكون لدى 50% من الأطفال المصابين صفات غير طبيعية عند الولادة (مثل حنف القدم أو التواء مشط القدم أو فلح حنكي أو أصابع مقوسة نحو الداخل بسبب عدم نمو العظام بشكل كامل أو قصر الأصابع، أو تورم الأذن الناجم عن الإصابة أثناء الولادة). يكون الطول ضمن المعدل الطبيعي قبل البلوغ. أما بعد البلوغ، يتضاءل نمو الأشخاص المصابين بـ MED كصفة متنحية لكن يبقون ضمن المعدل الطبيعي. قد يظهر التصوير الشعاعي الجانبي للركبة رضفة متعددة الطبقات.
الوراثة
يشمل خلل التنسج المشاشي المتعدد (MED) طيفًا من الاضطرابات الهيكلية، معظمها موروثة كصفة جسدية سائدة. ومع ذلك، يوجد شكل وراثي جسدي متنحٍ.[5]
تشمل الجينات المرتبطة COL9A1[6] و COL9A2[7] و COL9A3 و COMP[8] و MATN3.[9]
الأسباب
في الشكل السائد، توجد الطفرات الممرضة في خمسة جينات: COMP (الصبغي 19(،COL9A1 (الصبغي 6)، COL9A2 (الصبغي 1)،COL9A3 (الصبغي 20)، و MATN3 (الصبغي 2). ومع ذلك، في ما يقارب 10-20% من العينات التي تم تحليلها، لا يمكن تحديد طفرة في أي من الجينات الخمسة المذكورة أعلاه، ما يشير إلى مشاركة طفرات في جينات أخرى لم تحدد بعد في إمراضية MED السائد.[10]
تكون الطفرة في جين COMP في 70% من مرضى MED المؤكدين بالتحليل الجزيئي. توجد طفرات في الإكسونات التي تشفر تكرارات النوع الثالث (الإكسونات 8-14) والمجال C الطرفي (الإكسونات 15-19).[11] تكون الطفرات الأكثر شيوعًا في COL9A1 في الإكسونات 8-10، و COL9A2 في الإكسونات 2-4، وCOL9A3 في الإكسونات 2-4. إجمالاً، توجد تلك الطفرات لدى 10% من المرضى. لدى الـ 20% الأخرى من المصابين طفرات في جين MATN3، وكلها موجودة داخل الإكسون 2. وقد أوصت الشبكة الأوروبية لخلل التنسج الهيكلي بنظام الاختبار التالي:
- الدرجة 1: COMP (الإكسونات 10- 15) وMATN3 (الإكسون 2).
- الدرجة 2: COMP (الإكسونات 8-9 و16-19).
- الدرجة 3: COL9A1 (الإكسون 8)، COL9A2 وCOL9A3 (الإكسون 3).
تشارك جميع تلك الجينات في إنتاج المطرس خارج الخلوي (ECM). ما زال دور COMP غير واضح. فهو بروتين غير كولاجيني في المطرس خارج الخلوي.[12] يمكن للطفرات في هذا الجين أن تسبب خلل التنسج الضموري الكاذب (PSACH). وهو يلعب دورًا في سلامة الغضروف الهيكلية من خلال تفاعله مع بروتينات المطرس خارج الخلوي الأخرى ويمكن أن يكون جزءًا من تفاعل الخلايا الغضروفية مع المطرس. وهو مثبط قوي للموت المبرمج للخلايا الغضروفية ويمكن أن يكبت موت الخلايا المبرمج. له دور آخر وهو الحفاظ على تقلص خلايا العضلات الملساء الوعائية تحت تأثير التنبيهات الفيزيولوجية أو المرضية.[13]
منذ عام 2003، استخدمت الشبكة الأوروبية لخلل التنسج الهيكلي نظامًا عبر الإنترنت لتشخيص الحالات المحالة إلى الشبكة قبل إجراء تحليل الطفرات لدراسة الطفرات التي تسبب PSACH أو MED.[14]
COL9A1 وCOL9A2 وCOL9A3 هي مرمزات جينية للكولاجين نمط 9، وهو مكون للغضروف الزجاجي. قد يلعب بروتين MATN3 دورًا في تكوين الشبكات الخيطية خارج الخلية وفي نمو واستتباب الغضاريف والعظام.[15]
في النمط المتنحي، تحدث طفرة في جين DTDST، المعروف أيضًا باسم SLC26A2، لدى نحو 90% من المرضى، ما يسبب خلل التنسج الضموري. وهو بروتين سكري عابر للغشاء ناقل للكبريتات وله دور في عديد من أنواع خلل التنسج الغضروفي. وهو ضروري لسلفنة البروتيوغليكان وتنظيم المطرس.[16]
التشخيص
يجب أن يستند التشخيص إلى النتائج السريرية والشعاعية ويمكن إجراء تحليل جيني.[17]
العلاج
يجب أن يرى الطبيب أخصائي العظام الأفراد الذين يعانون من الأعراض ليقيم إمكانية العلاج (العلاج الفيزيائي لتقوية العضلات، والاستخدام الحذر للمسكنات مثل مضادات الالتهاب اللاستيرويدية). على الرغم من عدم وجود علاج، تستخدم الجراحة أحيانًا لتخفيف الأعراض.[18] قد تكون الجراحة ضرورية لعلاج خلل توضع الورك (قطع عظم الحوض أو عنق الفخذ) وفي بعض الحالات تشوه (على سبيل المثال الساق المقوسة أو الركبة الروحاء).[19] في بعض الحالات، قد يكون استبدال الورك الكلي ضروريًا. ومع ذلك، فإن الجراحة ليست ضرورية أو مناسبة دائمًا.[20]
يجب تجنب ممارسة الألعاب الرياضية التي تتضمن زيادة الحمل على المفاصل، ويُنصح بشدة بممارسة السباحة أو ركوب الدراجات.[21] يجب تجنب ركوب الدراجات عند الأشخاص الذين يعانون من ارتخاء الأربطة.
ينصح أيضًا بمراقبة الوزن.[22]
يعد استخدام العكازات أو غيرها من الوسائل المساعدة كالكرسي المتحرك مفيدًا لمنع آلام الفخذ.[23] يمكن تجنب الألم في اليد أثناء الكتابة باستخدام قلم ذو قبضة عريضة.[24]
التاريخ
أول من وصف خلل التنسج المشاشي المتعدد كان سيفيد ريبينغ وهارولد آرثر توماس فيربانك في ثلاثينيات القرن العشرين.
في عام 1994، حددت مجموعة رالف أولهمان ارتباط MED بالمنطقة المحيطة بالقسيم المركزي للصبغي 19، باستخدام تحليل الارتباط الوراثي.[25] ربطت مجموعة مايكل بريغز خلل التنسج الضموري الكاذب PSACH بنفس المنطقة.[26] رُبط جينCOMP أولاً بـ MED و PSACH في عام 1995.[27] في عام 1995، أجرت المجموعة التي يقودها نولتون بحثًا جينيًا وفيزيائيًا عالي الدقة لخلل التنسج المشاشي المتعدد وخلل التنسج الضموري الكاذب في الكروموزومين 19-p13 (الذراع القصيرة للصبغي 19 المنطقة 1 تحت المنطقة 3) و1-p12 (الذراع القصيرة للصبغي 1 المنطقة 1 تحت المنطقة 2).[28]
أدت البحوث التي أُجريت حول جين COMP لاستخدام نموذج الفأر لدراسة إمراضية MED. في عام 2002، أنتجت مجموعة سفينسون فأرًا يملك جين COMP معطلًا لدراسة البروتين COMP في الجسم الحي. لم تُظهر هذه الفئران أي تشوهات تشريحية أو نسيجية أو حتى تشوهات في البنية الداخلية ولم تظهر أي علامات سريرية تشير إلى PSACH أو MED. لم يُعوض نقص COMP بواسطة أي بروتين آخر في عائلة بروتين الثرومبوسبوندين. أكدت هذه الدراسة أن هذا المرض لا ينجم عن انخفاض في التعبير عن COMP.[29]
في عام 2007، أنتجت مجموعة بيروغ غارسيا نموذجًا آخر لفأر يحمل طفرة كانت موجودة سابقًا لدى مريض بشري. مع هذا النموذج الجديد، تمكنوا من إثبات أن انخفاض تكاثر الخلايا وزيادة موت الخلايا المبرمج هي آليات مرضية هامة تشارك في حدوث MED وPSACH.[30] في عام 2010، سمح نموذج الفأر هذا بإلقاء نظرة جديدة على الاعتلال العضلي والاعتلال الوتري، واللذين يرتبطان غالبًا بـ PSACH وMED. أظهر هؤلاء المرضى زيادة في إجهاد العضلات والهيكل العظمي، والذي اتضح من الزيادة في الألياف العضلية ذات النواة المركزية. ينتج الاعتلال العضلي لدى الفأر الطافر عن اعتلال الأوتار الأساسي. توجد نسبة أعلى من ألياف الكولاجين ذات القطر الأكبر، لكن المساحة المستعرضة للأوتار الطافرة بأكملها كانت أيضًا أقل بكثير من تلك الموجودة في الأوتار الطبيعية ما يسبب كزازة المفصل وتيبسه وبالتالي الضعف وسهولة التعب. هذه الدراسة مهمة لأن هذه الأمراض غالبًا ما يتم خلطها بالمشاكل العصبية، إذ يمكن للطبيب اكتشاف ضعف العضلات. وهذا يشمل العديد من الفحوص العصبية السريرية المؤلمة وغير المجدية قبل إعطاء التشخيص الصحيح. لهذا السبب، اقترح الباحثون على طبيب الأطفال إجراء الأشعة السينية قبل بدء التقييم العصبي، لاستبعاد خلل التنسج.[31]
اكتُشفت طفرة COL91A في عام 2001.[32]
المراجع
- Disease Ontology (بالإنجليزية), 27 May 2016, QID:Q5282129
- EL-Sobky، TA؛ Shawky، RM؛ Sakr، HM؛ Elsayed، SM؛ Elsayed، NS؛ Ragheb، SG؛ Gamal، R (15 نوفمبر 2017). "A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review". J Musculoskelet Surg Res. ج. 1 ع. 2: 25. DOI:10.4103/jmsr.jmsr_28_17.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link) - Canepa، Giuseppe؛ Maroteaux، Pierre؛ Pietrogrande، Vincenzo (2001). Dysmorphic-syndromes and constitutional diseases of the skeleton. Padova: Piccin. ISBN:978-88-299-1502-6.
- Lachman، RS؛ Krakow، D؛ Cohn، DH؛ Rimoin، DL (2005). "MED, COMP, multilayered and NEIN: an overview of multiple epiphyseal dysplasia". Pediatr Radiol. ج. 35 ع. 2: 116–23. DOI:10.1007/s00247-004-1323-4. PMID:15503005.
- "Multiple Epiphyseal Dysplasia (MED) - Pediatrics - Orthobullets". مؤرشف من الأصل في 2018-05-05.
- "COL9A1 collagen type IX alpha 1 [ Homo sapiens (human) ]". مؤرشف من الأصل في 2019-09-11.
- "COL9A2 collagen type IX alpha 2 [ Homo sapiens (human) ]". مؤرشف من الأصل في 2019-09-11.
- "COL9A3 collagen type IX alpha 3". مؤرشف من الأصل في 2019-03-28.
- "MATN3 matrilin 3 [ Homo sapiens (human) ]". مؤرشف من الأصل في 2019-03-28.
- d Briggs، Michael؛ Wright، Michael J؛ Mortier، Geert R (25 يوليو 2013) [2003]. "Multiple Epiphyseal Dysplasia, Dominant - GeneReviews® - NCBI Bookshelf". University of Washington, Seattle. PMID:20301302. مؤرشف من الأصل في 2014-05-03. اطلع عليه بتاريخ 2014-05-03.
{{استشهاد بدورية محكمة}}: الاستشهاد بدورية محكمة يطلب|دورية محكمة=(مساعدة) والوسيط|الفصل=تم تجاهله (مساعدة) - Briggs MD، Chapman KL (2002). "Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations". Hum Mutat. ج. 19 ع. 5: 465–78. DOI:10.1002/humu.10066. PMID:11968079.
- Paulsson M، Heinegård D (1981). "Purification and structural characterization of a cartilage matrix protein". Biochem J. ج. 197 ع. 2: 367–75. DOI:10.1042/bj1970367. PMC:1163135. PMID:7325960.
- "GeneCards". مؤرشف من الأصل في 2017-01-13.
- Jackson GC، Mittaz-Crettol L، Taylor JA، Mortier GR، Spranger J، Zabel B، وآخرون (2012). "Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution". Hum Mutat. ج. 33 ع. 1: 144–57. DOI:10.1002/humu.21611. PMC:3272220. PMID:21922596.
- "MATN3 review". مؤرشف من الأصل في 2010-12-05.
- "SLC26A2 solute carrier family 26". مؤرشف من الأصل في 2010-12-05.
- Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993.
- Trehan R، Dabbas N، Allwood D، Agarwal M، Kinmont C (2008). "Arthroscopic decompression and notchplasty for long-standing anterior cruciate ligament impingement in a patient with multiple epiphyseal dysplasia: a case report". J Med Case Reports. ج. 2: 172. DOI:10.1186/1752-1947-2-172. PMC:2412893. PMID:18498631.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link) - Linden، Suzanne K. Campbell, Robert J. Palisano, Darl W. Vander (2005). Physical therapy for children (ط. 3rd). Philadelphia, Pa.: Elsevier Saunders. ISBN:978-0-7216-0378-0.
{{استشهاد بكتاب}}: صيانة الاستشهاد: أسماء متعددة: قائمة المؤلفين (link) - Bajuifer S، Letts M (أبريل 2005). "Multiple epiphyseal dysplasia in children: beware of overtreatment!" (PDF). Can J Surg. ج. 48 ع. 2: 106–9. PMC:3211605. PMID:15887789. مؤرشف من الأصل (PDF) في 7 ديسمبر 2018. اطلع عليه بتاريخ أكتوبر 2020.
{{استشهاد بدورية محكمة}}: تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Juergen Maeurer (2006). Imaging strategies for the knee. ISBN:978-3-13-140561-6.
- Paans N، van den Akker-Scheek I، van der Meer K، Bulstra SK، Stevens M (2009). "The effects of exercise and weight loss in overweight patients with hip osteoarthritis: design of a prospective cohort study". BMC Musculoskelet Disord. ج. 10: 24. DOI:10.1186/1471-2474-10-24. PMC:2649885. PMID:19236692.
{{استشهاد بدورية محكمة}}: صيانة الاستشهاد: دوي مجاني غير معلم (link) - L.Echternach، Ed.John (1990). Physical therapy of the hip. New York...[etc.]: Churchill Livingstone. ISBN:978-0-443-08650-2.
- Children's Orthopaedics and Fractures. Springer. 2 فبراير 2010. ISBN:978-1-84882-610-6. مؤرشف من الأصل في 2014-07-06. اطلع عليه بتاريخ 2014-05-03.
{{استشهاد بكتاب}}: الوسيط غير المعروف|المحررين=تم تجاهله (مساعدة) - Oehlmann R، Summerville GP، Yeh G، Weaver EJ، Jimenez SA، Knowlton RG (1994). "Genetic linkage mapping of multiple epiphyseal dysplasia to the pericentromeric region of chromosome 19". Am J Hum Genet. ج. 54 ع. 1: 3–10. PMC:1918067. PMID:8279467.
- Briggs MD، Rasmussen IM، Weber JL، Yuen J، Reinker K، Garber AP، وآخرون (1993). "Genetic linkage of mild pseudoachondroplasia (PSACH) to markers in the pericentromeric region of chromosome 19". Genomics. ج. 18 ع. 3: 656–60. DOI:10.1016/S0888-7543(05)80369-6. PMID:8307576.
- Briggs MD، Hoffman SM، King LM، Olsen AS، Mohrenweiser H، Leroy JG، وآخرون (1995). "Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene". Nat Genet. ج. 10 ع. 3: 330–6. DOI:10.1038/ng0795-330. PMID:7670472.
- Knowlton RG، Cekleniak JA، Cohn DH، Briggs MD، Hoffman SM، Brandriff BF، وآخرون (1995). "High-resolution genetic and physical mapping of multiple epiphyseal dysplasia and pseudoachondroplasia mutations at chromosome 19p13.1-p12". Genomics. ج. 28 ع. 3: 513–9. DOI:10.1006/geno.1995.1183. PMID:7490089.
- Svensson L، Aszódi A، Heinegård D، Hunziker EB، Reinholt FP، Fässler R، وآخرون (2002). "Cartilage oligomeric matrix protein-deficient mice have normal skeletal development". Mol Cell Biol. ج. 22 ع. 12: 4366–71. DOI:10.1128/mcb.22.12.4366-4371.2002. PMC:133870. PMID:12024046.
- Piróg-Garcia KA، Meadows RS، Knowles L، Heinegård D، Thornton DJ، Kadler KE، وآخرون (2007). "Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP". Hum Mol Genet. ج. 16 ع. 17: 2072–88. DOI:10.1093/hmg/ddm155. PMC:2674228. PMID:17588960.
- Piróg KA، Jaka O، Katakura Y، Meadows RS، Kadler KE، Boot-Handford RP، وآخرون (2010). "A mouse model offers novel insights into the myopathy and tendinopathy often associated with pseudoachondroplasia and multiple epiphyseal dysplasia". Hum Mol Genet. ج. 19 ع. 1: 52–64. DOI:10.1093/hmg/ddp466. PMC:2792148. PMID:19808781.
- Czarny-Ratajczak M، Lohiniva J، Rogala P، وآخرون (نوفمبر 2001). "A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity". Am. J. Hum. Genet. ج. 69 ع. 5: 969–80. DOI:10.1086/324023. PMC:1274373. PMID:11565064.
 بوابة طب
بوابة طب