ابيضاض الدم الليمفاوي المزمن
ابيضاض الدم الليمفاوي المزمن (بالإنجليزية: Chronic Lymphocytic Leukemia) هو مرض سرطاني يصيب الخلية الليمفية من نوع B.[1][2][3] يصيب البالغين وخاصة الأفراد التي تتراوح أعمارهم ما بين 60- 80 عاما خاصة في أوروبا وأميركا الشمالية، وينتشر بين الذكور بنسبة أعلى من الإناث (1:2). يشكل ما نسبته 25% من الإصابات بمختلف أمراض أبيضاض الدم. في أميركا الشمالية، معدل الإصابة السنوي للرجال 3.35-3.69 لكل 000 100 شخص، أما بالنسبة للنساء هو 1.61-1.92 لكل 000 100 شخص. يتصف المرض بالتراكم التصاعدي للخلايا الليمفية في نخاع العظم والعقد الليمفية، وبالتالي انتقال المرض تدريجيا إلى بقية الأعضاء المصنعة للدم. يتم إيعاز المرض إلى خلل جزيئي يؤدي إلى تأخير أو إعطاب عملية استموات الخلايا.
| ابيضاض الدم الليمفاوي المزمن | |
|---|---|
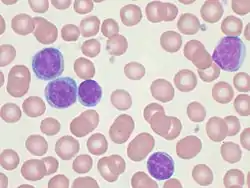 مسحة الدم المحيطي تظهر خلايا CLL مسحة الدم المحيطي تظهر خلايا CLL | |
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | ابيضاض الدم المزمن، ولمفوما، وابيضاض الدم، ومرض |
| الإدارة | |
| أدوية | يوراموستين، وأليمتوزوماب، ومركابتوبورين، وسيكلوفوسفاميد، وكلورامبوسيل، وبيغاسبارغاس، وكلادريبين، وإيفوسفامايد، وبريدنيزون، وبينداميوستين، وليناليدوميد، وبنتوستاتين، ونيلوتينيب، وبيغفيلغراستيم، ودوكسوروبيسين، وإبروتينيب، وديكساميثازون، وفينيتوسلاكس، وفينكريستين، وريتوكسيماب، وأوفاتوموماب، وأوبينوتوزوماب، وكلورامبوسيل، وسيكلوفوسفاميد، وفينيتوسلاكس
|
أسباب المرض
بخلاف بقية أنواع أبيضاض الدم، لم يتم ربط الإصابة بالمرض مع عوامل مخطرة بيئية أو التعرض لأي مركبات كيميائية أو أشعة. أحد العوامل المخطرة المرض هو وجود إصابة بهذا المرض أو أية اضطرابات تكاثر ليمفي في أحد أفراد العائلة، وجد أن مصاب من كل 10 أشخاص مصابين بأبيضاض الدم الليمفي المزمن لديهم إصابة عائلية بالمرض.
الفسيولوجيا المرضية
وجد الباحثون أن من ميزات المرض هو تراكم وتكدس الخلايا الليمفية وليس من الانقسام الغير طبيعي للخلايا. ومن هنا تم ربط المرض بعملية استموات الخلايا. تتم عملية استموات الخلايا من خلال طريقين رئيسين هما الطريق الداخلي (ميتكوندريا) والطريق الخارجي (TNF). تتضمن عمليات نقل إشارة وتنشيط وتثبيط مركبات معينة تؤدي بالنهاية لتحفيز عملية استموات الخلايا.
في أبيضاض الدم الليمفي المزمن تتدخل عدة بروتينات في الطريقين الداخلي والخارجي مما يؤدي إلى تثبيط عملية استماتة الخلايا. مثل هذه البروتينات عائلة بي سي إل 2 و NF-KB و Akt. ووجد أيضا أن بعض الاضطرابات الوراثية الخلوية تؤدي إلى إخلال في عمل المورثة بي53 المسؤولة عن تثبيط الأورام.
الأعراض
معظم حالات اكتشاف المرض تتم بالصدفة من خلال الفحوصات الروتينية أو بسبب ظهور أعراض فقر الدم أو تضخم متماثل في الغدد الليمفاوية وخاصة التي تقع في منطقة العنق والإبط، تظهر 3/2 الحالات تضخم في الكبد أوالطحال. وتظهر بعض الحالات تضخم في اللوزتين وبعض الكدمات لقلة الصفائح الدموية.
العلامات المخبرية
تظهر الفحوصات المخبرية ارتفاع في أعداد كريات الدم البيضاء الليمفية ووجودها بما نسبته 40% أو أكثر في نخاع العظم. في صورة الدم، تظهر الخلايا بأحجام صغيرة وكمية السايتوبلازم تكون ضئيلة ولا تظهر النوية بشكل واضح.
يشاهد في صورة الدم خلايا تعرف باسم سلائف الليمفيات، وتكون كبيرة الحجم مقارنة بالكريات الليمفية ولا تزيد عن 5%. في بعض الحالات المرضية تشاهد بنسبة أعلى، في تلك الحالة يتم تشخيص الحالة كنوع من أنواع المرض ويسمى أبيضاض الدم الليمفي المزمن/سلائف الليمفيات.
فحص نقي العظم
يشكل فحص نقي العظم أهمية كبيرة في عملية تشخيص المرض، ومتابعة العلاج. تكمن أهميته في النقاط التالية:
- وسيلة لتحديد مآل ومرحلة المرض: عندما يكون نخاع العظم مكدس بالخلايا الليمفية مع عديد قليل جدا من المناطق الدهنية (النمط المنتشر)، هذا يؤشر إلى أن المرض في مرحلة متأخرة وبالعادة يكون مآل المرض سيئ. وقد يكون نمط نخاع العظم خلالي مع وفرة المساحات الدهنية أو يكون نمط عقدي الشكل. بعض الإصابات تظهر مزيج من النمطين الأخيرين.
- تحديد طبيعة ومسبب قلة الحُمُر والصفائح: يساعد فحص نخاع العظم على تحديد مسبب قلة الخلايا في الدم وبالتالي معالجة المسبب.
- التشخيص التفريقي مع الأمراض الأخرى: يتم ملاحظة نمط ترشيح الخلايا الليمفية يساعد على تشخيص أبيضاض الدم الليمفي المزمن من بقية الأمراض التي تتشابه معه.
- متابعة واستجابة المريض للعلاج.
مآل المرض
هناك عدة عوامل لها دور في تحديد مآل المرض.
| عوامل محددة لمآل المرض | |||
| العامل | جيد | سيئ | |
|---|---|---|---|
| مرحلة المرض | بينت A راي (0-I) | بينت B,C راي (II-IV) | |
| الجنس | انثى | ذكر | |
| مدة تضاعف الليمفيات | بطيئ | سريع | |
| نمط خزعة نخاع العظم | عقدي | منتشر | |
| الوراثة الخلوية | حذف 13q4 | حذف 17p، حذف 11q23، تثلث صبغي 12 | |
| مورثة VH | مطفرة | طبيعية | |
| تعبير CD38 | قليل | مرتفع | |
| تعبير ZAP-70 | قليل | مرتفع | |
أنظمة تحديد مرحلة المرض
هنالك نظامان يستخدمان لتحديد مرحلة المرض، النظام الأول، نظام راي (1975)، قسمت مجموعة راي المرض لعدة مراحل:[4]
- مرحلة 0: تتصف بارتفاع كريات الدم البيضاء الليمفاوية فقط
- مرحلة I: تضخم في الغدد الليمفاوية
- مرحلة II: تضخم في الكبد أو الطحال أو كلاهما
- مرحلة III: فقر الدم
- مرحلة IV: هبوط في الصفائح
النظام الآخر هو نظام بينت (1981).[5] طور بينت هذا النظام بالاعتماد على دراستين فرنسيتين. يتم تقسيم مراحل المرض بالاعتماد على عدد الأعضاء اللمفاوية المتضخمة (العنق والإبط والمنطقة الأربية والكبد والطحال) كالتالي:
- A: ارتفاع كريات الدم البيضاء الليمفاوية فقط مع عدم وجود أي تضخم أو تضخم عضوين أو أقل
- B: تضخم 3 أعضاء أو أكثر
- C: وجود فقر دم أوانخفاض في الصفائح الدموية
الوراثة الخلوية
أظهرت الأبحاث أن ما نسبته 80% على الأقل من حالات الإصابة بالمرض تحوي على اضطراب وراثي خلوي أو أكثر. يتم تشخيص هذه الاضطرابات بواسطة فحص التهجين الموضعي المتألق، لما لها من دور في تحديد مآل المرض. صدرت دراسة في عام 2000 قام بها دونر ومجموعته بدراسة 325 مريض وخرجوا بمجموعة من الاضطرابات الوراثية الخلوية ورتبوها حسب دورها في مآل المرض من السيئ إلى الجيد.
| الاضطرابات الوراثية الخلوية حسب دراسة دونر مرتبة حسب دورها في مآل المرض | |||
| الاضطرابات الوراثية الخلوية | نسبة الإصابة | ||
|---|---|---|---|
| حذف 17p | 7% | ||
| حذف 11q | 17% | ||
| تثلث صبغي 12 | 14% | ||
| نمط نووي طبيعي | 18% | ||
| حذف 13q4 | 36% | ||
| غيرها | 8% | ||
مورثة VH
كان الاعتقاد السائد أن المرض يحدث في الخلايا الليمفية التي تحوي نسخة طبيعية من مورثة VH. إلا أنه اكتشف حديثا أن أكثر من نصف حالات المرضى (55%) يحملون مورثة VH مُطفّرة. تكمن أهمية حدوث هذه الطفرة لتعزز قدرة الخلايا الليمفية على إنتاج أضداد متخصصة. وجدت الدراسات أن الأشخاص الذين يحملون النسخة المطفرة من المورثة يكون مآل المرض أفضل من المرضى الذين يحملون النسخة الطبيعية. يتم التحري عن هذه الطفرة إما بقياس كميات CD38 في الخلايا الليمفية أو التحري عن التعبير الجيني لمورثة ZAP-70. كلاهما ينتج بكميات أكبر في حالة المورثة الطبيعية.
التشخيص التفريقي
تشتبه بعض الأمراض في الأعراض والظواهر مما تجعل مهمة التشخيص صعبة في بعض الأحيان مثل هذه الأمراض متلازمة ريختر وأبيضاض الدم الليمفي المزمن العائلي وكثرة الليمفيات البائية الوحيدة ومتعددة النسيلة.
تم تطوير نظام نقاط حتى يتم التفريق بين أبيضاض الدم الليمفي المزمن وغيره وذلك بالاعتماد على وجود بعض المؤشرات. هناك 5 مؤشرات يتم التحري عنها بواسطة جهاز عداد الخلايا. كل منها يشكل نقطة واحدة. تحسب النقاط بعدها ويتم تشخيص المرض بالاعتماد على تلك المؤشرات.
العلاج
تتم معالجة المرض بعدة طرق:
- العلاج الكيميائي: ويستخدم للقضاء على الخلايا السرطانية ومنعها من الانقسام مثل كلورامبيوسيل (Chlorambucil) وفلودارابين (Fludarabine).
- العلاج الإشعاعي: يتم تعريض المريض للأشعة العلاجية لتقليص حجم الغدد الليمفية والتي لا تستجيب للعلاج الكيميائي.
- الأضداد وحيدة النسيلة: يتم استخدام الريتوكسيماب (Rituximab) للقضاء على الخلايا السرطانية.
- زراعة نخاع العظم.
المراجع
- Venetoclax label. fda.gov نسخة محفوظة 16 فبراير 2017 على موقع واي باك مشين.
- Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS (أغسطس 1975). "Clinical staging of chronic lymphocytic leukemia". Blood. ج. 46 ع. 2: 219–34. PMID:1139039.
- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK (1999). "Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia". Blood. ج. 94 ع. 6: 1848–54. PMID:10477713. مؤرشف من الأصل في 2010-07-22.
- Rai، K. R.؛ Sawitsky، A.؛ Cronkite، E. P.؛ Chanana، A. D.؛ Levy، R. N.؛ Pasternack، B. S. (1975-08). "Clinical staging of chronic lymphocytic leukemia". Blood. ج. 46 ع. 2: 219–234. ISSN:0006-4971. PMID:1139039. مؤرشف من الأصل في 30 سبتمبر 2022.
{{استشهاد بدورية محكمة}}: تحقق من التاريخ في:|تاريخ=(مساعدة) - Binet، J. L.؛ Leporrier، M.؛ Dighiero، G.؛ Charron، D.؛ D'Athis، P.؛ Vaugier، G.؛ Beral، H. M.؛ Natali، J. C.؛ Raphael، M. (1977-08). "A clinical staging system for chronic lymphocytic leukemia: prognostic significance". Cancer. ج. 40 ع. 2: 855–864. DOI:10.1002/1097-0142(197708)40:2<855::aid-cncr2820400239>3.0.co;2-1. ISSN:0008-543X. PMID:890666. مؤرشف من الأصل في 30 سبتمبر 2022.
{{استشهاد بدورية محكمة}}: تحقق من التاريخ في:|تاريخ=(مساعدة)
 بوابة طب
بوابة طب